
Introduction
Systemic sclerosis (SSc) is a chronic autoimmune disease which still poses a great challenge to clinicians. The most prominent feature of SSc is the process of progressive fibrosis resulting from the excessive deposition of extracellular matrix components in different tissues and organs. Vascular damage, inflammation and the presence of specific autoantibodies are also characteristic for SSc [1]. Systemic sclerosis affects skin and internal organs, such as lungs, heart, kidneys, musculoskeletal system and the gastrointestinal tract. Skin sclerosis is a main symptom of SSc, evaluated using the modified Rodnan skin score (mRss) and an easily detectable marker of disease activity [2].
Systemic sclerosis is classified into two subsets based on the extent of skin involvement – limited systemic sclerosis (lcSSc) and diffuse systemic sclerosis (dsSSc) [3]. Patients with fibrosis of the skin affecting acral parts of the body – face and limbs (distal to the knees and elbows) – are classified as having lcSSc, whereas those with fibrosis of the trunk and proximal parts of the limbs are classified as having dsSSc [3, 4]. Although lcSSc has slow progression of skin fibrosis and Raynaud’s phenomenon starts long before the skin symptoms, it is not limited to skin involvement and is also associated with the involvement of the esophagus and lungs [4]. However, late and slow organ involvement in lcSSc is associated with relatively good prognosis with 10-year survival over 90% [5]. Patients with dsSSc have poorer prognosis because of fast progression of skin and organ involvement including cardiovascular system, lungs, kidneys, gastrointestinal tract and even central and peripheral nervous system. In dsSSc there is a shorter time period between the onset of Raynaud’s phenomenon and skin symptoms, although there are studies contradicting this conclusion [6, 7]. The overall 10-year survival rate in dsSSc ranges from 65 to 82%, which is a result of the wide range of systemic complications [5]. The most life-threatening of them affect the heart, lungs and kidneys [3, 8, 9].
In systemic sclerosis sine scleroderma (ssSSc), affecting 5% of patients with SSc, there are typical SSc symptoms (positive autoantibodies, Raynaud’s phenomenon, involvement of the lungs), but no skin fibrosis is present [10].
In 2013 the American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR) published new classification criteria and guidelines for SSc management [11]. As the knowledge about SSc pathogenesis broadens, factors that cause or influence the development of fibrosis, such as infections, chemical (silica, vinyl chloride) and physical factors are still being analyzed. Their activation is associated with genetic susceptibility correlated with the presence of certain susceptibility genes: HLA (e.g. A23, B18 or DR11) and non-HLA (e.g. CD247, signal transducer and activator of transcription protein 4 (STAT4) and interferon regulatory factor 5 (IRF5), B cell scaffold protein with ankyrin repeats gene (BANK1) [12], variants of autophagy protein 5 (ATG5) or c-SRC tyrosine kinase (CSK) [13]. Epigenetic modifications that play a role in SSc pathogenesis include DNA demethylation of eNOS, CD40L and CD70 (regulatory genes).
Currently special attention is paid to the role of regulatory molecules, such as microRNA (miRNAs), in the development and course of autoimmune diseases [14]. Only the coincidence of these factors creates a risk of activation of innate and acquired immune response in susceptible subjects. The deficiency of vitamin D is believed to be important in the development of SSc. Recent research showed that also factors such as lysophosphatidic acid (LPA) or sphingosine 1-phosphate (S1P) play a role in the vascular wall damage and fibrosis in SSc [15].
Autoantibodies in systemic sclerosis
The immune system activation in SSc results in production of autoantibodies. Antinuclear antibodies (ANA) are the most frequently detected autoantibodies in patients with SSc (90–95% of cases). In the case of patients with clinical symptoms of SSc but without circulating ANA the differential diagnosis of scleroderma-like diseases is obligatory. In SSc there are several disease-specific autoantibodies associated with different types of the disease, also determining the prognosis. Autoantibodies against topoisomerase I (Topo I, ATA, Scl-70) are more often observed in patients with dcSSc and they are associated with pulmonary complications, digital ulcers and progression of hand disability [16]. In lcSSc anti-centromere antibodies (ACA) are commonly detected and they increase the risk of pulmonary fibrosis and pulmonary hypertension. In some patients with dcSSc antibodies against RNA polymerase III are detected, carrying a higher risk of a renal crisis [16, 17]. They are also present in patients with concomitant neoplastic process. Antibodies against fibrillarin (anti-U3RNP) and against Th/To occur less frequently. The former are associated with pulmonary arterial hypertension and cardiovascular complications, and the latter are characteristic for lcSSc [16–18]. Table I summarizes autoantibodies in SSc.
Table I
Autoantibodies specific and associated with systemic sclerosis (SSc)
Skin involvement
The term “scleroderma” derives from the most characteristic feature of the disease – skin thickening [19]. In the diffuse subtype of SSc (dcSSc) Raynaud’s phenomenon occurs simultaneously with skin thickening or shortly prior to it. The dermal induration is commonly associated with hypo- or hyperpigmentation and telangiectasias of the skin, and can be observed on the trunk, extremities, neck and face, giving typical sharp facial features called “mask-like face”. The nasal alar and red lips are thinned and radial furrowing around the mouth is present. In more severe cases eyelid movement can be limited and the aperture of the mouth is narrowed (microstomy). In early stage of dcSSc fingers are swollen (puffy fingers) and progressively skin becomes indurate and thin, especially on fingertips and nail protuberances, where hard-to-heal erosions and ulcers occur rapidly. The skin thickening decreases proximally. With the progression of the disease finger contractures (sclerodactyly) develop. The range of finger motion is limited, while trophic disorders contribute to bone destruction and shortening of distal phalanges (acroosteolysis). As the function of sudoriferous and sebaceous glands is impaired, dryness of the conjunctiva and oral mucosa is commonly observed, and less frequently xerosis cutis and alopecia [19, 20].
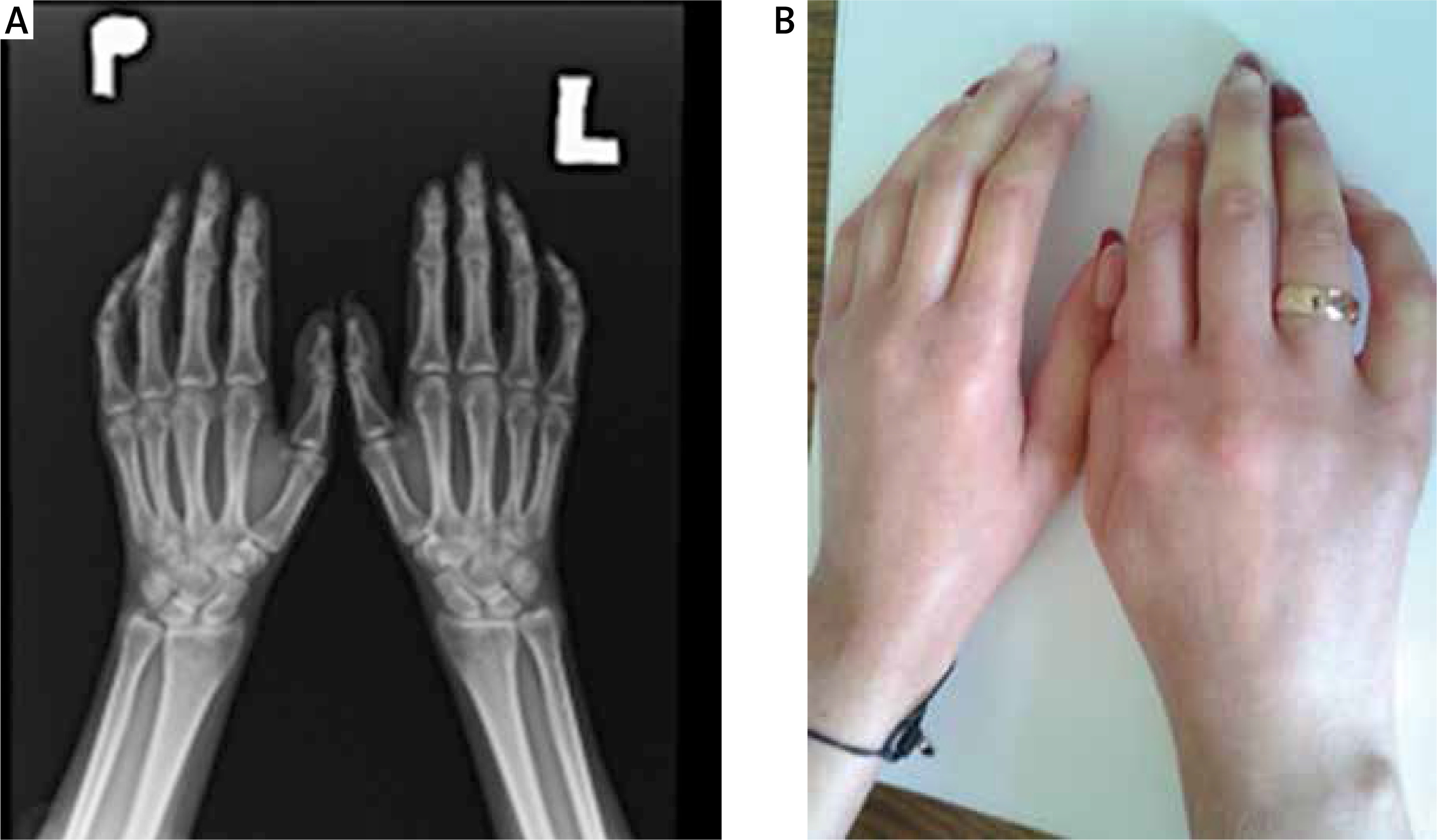
Limited systemic sclerosis progresses slowly and the microcirculation disturbances, such as Raynaud’s phenomenon, are usually ahead of dermal induration of the face and proximal parts of upper extremities. Skin lesions in both subtypes of SSc are similar, but telangiectasias in lcSSc are more advanced, while pigmentation disorders are not as prominent as in dcSSc. Infrequently, as an effect of calcium deposition in the skin, hard subcutaneous nodules around small joints of the hand can appear (calcinosis cutis). On radiographs calcinosis in soft tissue around the joints – especially of the fingers – may be present (Fig. 1). The course of SSc sine scleroderma is similar to lcSSc.
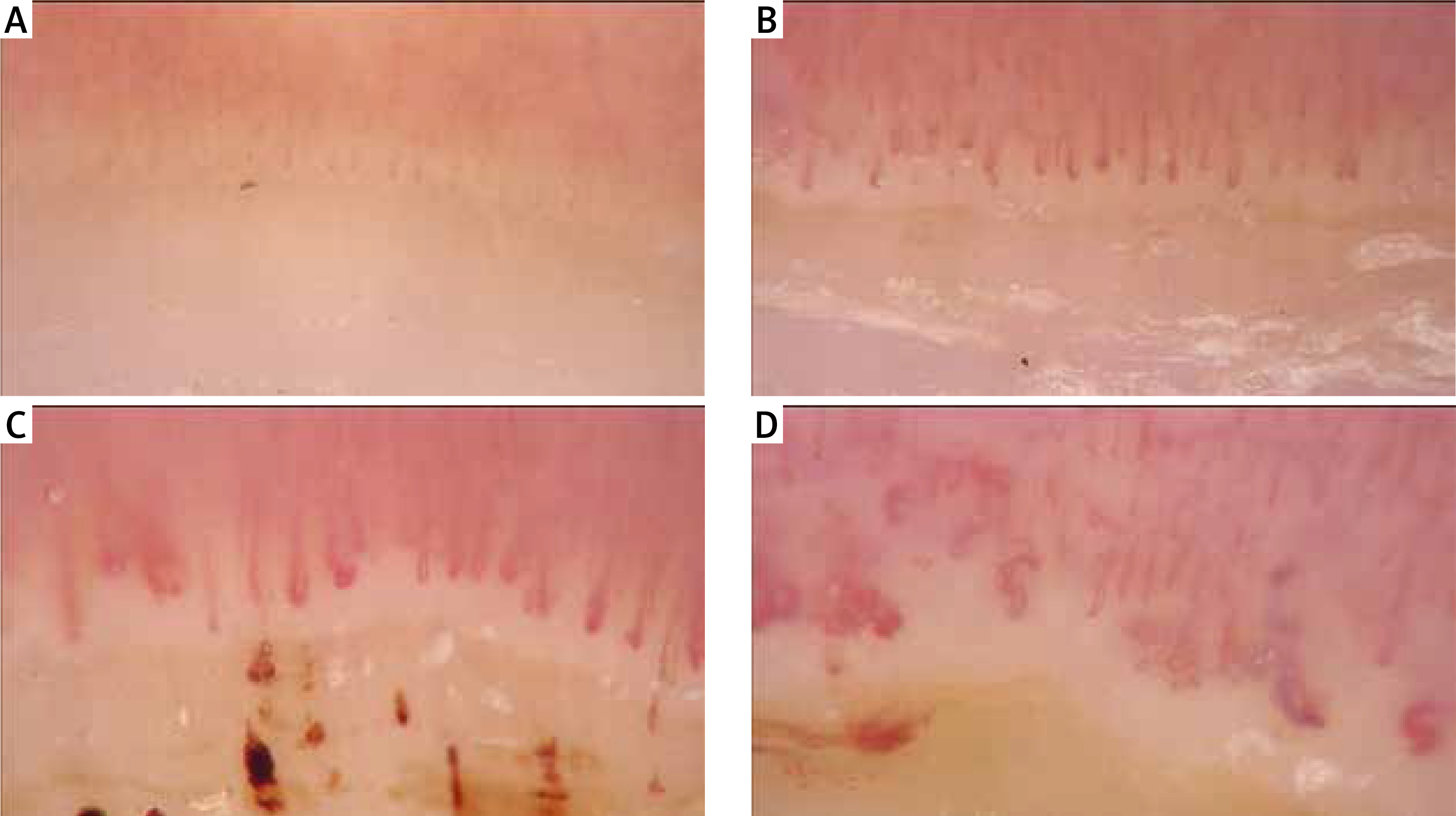
Systemic sclerosis is characterized by specific capillaroscopy changes – with patterns of early and late vascular changes (Fig. 2) [8, 21].
Fig. 2
Images of nailfold capillaroscopy from normal image (A), early (B), active (B) to late stage (D) with giant capillaries and avascular areas.
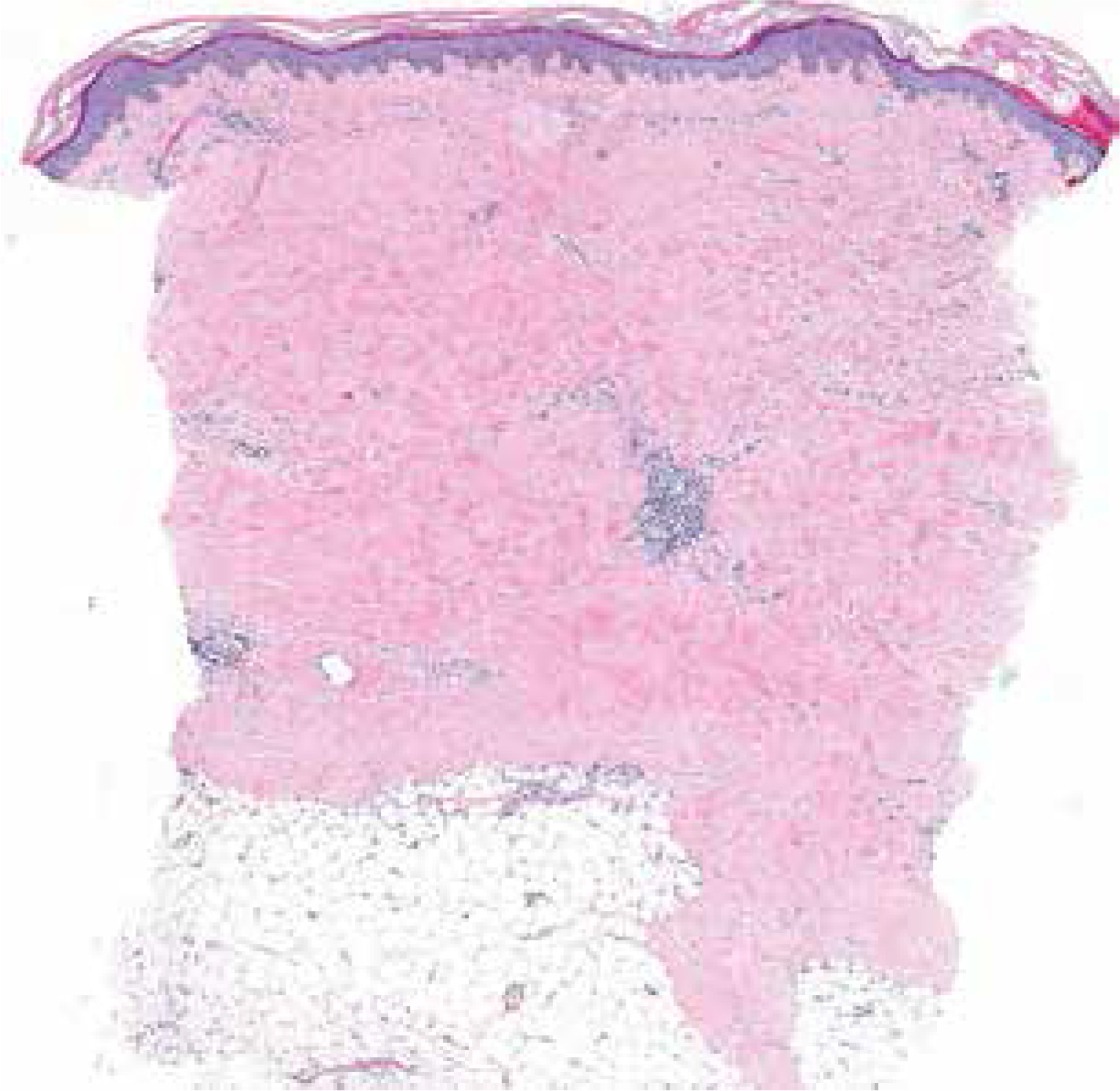
The histopathological assessment is not recommended in routine practice as clinical features are generally sufficient to make a proper diagnosis. In unclear and diagnostically difficult cases a skin biopsy may be performed (Fig. 3), revealing atrophic areas and increased collagen deposition with diminution of the spaces between normal collagen bundles. Encroachment around hair follicles and eccrine units with loss of periadventitial fat may be encountered and is characteristic for SSc [22].
Gastrointestinal tract
Gastrointestinal tract (GIT) involvement is common (90%) in patients with SSc [23], affecting all parts of the GIT. Within the oral cavity the most frequent complications of SSc are microstomia and xerostomia. The latter can be an effect of a concomitant Sjögren’s syndrome. A panoramic radiograph should be performed in every scleroderma case; radiographic changes include mainly osteolysis of the mandibular arch and coronoid process and are related to the tension of the facial skin, microstomia and the atrophy of masseter and pterygoid muscles [24]. The esophagus is the most frequently involved organ of GIT in SSc – almost 67% of patients complaint of one or more symptoms: dysphagia, odynophagia, regurgitation, pyrosis, chronic cough or hoarseness [25]. They are a result of structural and functional alterations of the esophagus, such as amyotonia contributing to dilation of the lumen, dysfunction of the lower esophageal sphincter (LES) and hiatal hernia – which lead to the increased incidence of gastroesophageal reflux disease (GERD) in SSc. Additionally, reduced LES pressure may lead to chronic reflux changes such as strictures and Barrett’s esophagus. The predominant alteration of the stomach in SSc is gastric antral vascular ectasia (GAVE). It can be easily diagnosed by gastroscopy, which reveals multiple parallel longitudinal columns of red vessels giving the distinctive “watermelon stomach” image. The symptoms of GAVE are iron deficiency anemia caused by chronic micro-bleeding or even acute GIT bleeding [26].
Gastroparesis is a functional disorder of the stomach which is observed in 50% of SSc cases and is confirmed in barium X-rays, scintigraphy or antroduodenal manometry. Some SSc patients present such symptoms as vomiting, diarrhea, and constipation, which suggests intestine involvement. The colonoscopy, radiographs and scintigraphy allow one to visualize such changes as the dilation of the bowel’s lumen, diverticuli and pneumatosis cystoides intestinalis. In the late stage of scleroderma diarrhea and undernutrition are frequently reported – as an effect of malabsorption and small intestinal bacterial overgrowth (SIBO). When the dysmotility affects the large intestine chronic intestinal pseudo-obstruction (CIPO) can occur – a rare complication mimicking the true bowel obstruction by giving similar symptoms: nausea, vomiting, abdominal distention and changes in bowel movement [27]. Anorectum involvement is the second most frequent (50–70%) alteration in GIT in SSc, resulting in complaints of fecal incontinence, constipation and rectal prolapse [28].
Nervous system
Central and peripheral nervous system involvement poses many diagnostic challenges. It has been previously assumed that in SSc changes in the nervous system are rare or secondary to other organ changes, such as vasculitis, kidney involvement, hypoxemia, pulmonary and systemic hypertension or associated with the therapy, especially with glucocorticosteroids. Recent reports suggest that the nervous system (NS) changes in SSc may be directly related to the disease [29]. It is assumed that the same vascular lesions as in Raynaud’s phenomenon may occur in the brain and peripheral small vessels vascularizing peripheral nerves, leading to symptoms of nervous system involvement [30].
The most common NS symptoms include headache and dizziness, convulsions, visual disturbances and aphasia. Less common symptoms include anxiety, depression, psycho-organic syndromes, disturbances of cognition and orientation, and even loss of consciousness.
Transient ischemic attack (TIA) and/or other ischemic syndromes, as well as strokes, trigeminal neuroinflammation, peripheral polyneuropathy and cranial nerve inflammation, also may be observed. Clinical symptoms depend on the stage of the disease. In the early stages, episodes of ischemic strokes, TIA or cerebral hemorrhages were observed, while further disease duration brings memory and mental disorders, as well as depressive episodes [31].
In the diagnosis of NS damage in SSc, neuroradiological methods such as magnetic resonance imaging (MRI), computed tomography (CT), single photon emission computed tomography (SPECT) and positron-emission tomography (PET) are used [32]. The nerve conduction studies are the principal diagnostic techniques to confirm peripheral nerve involvement. Electromyography (EMG) allows one to demonstrate the nature of peripheral nerve involvement, to differentiate between neurogenic and myogenic lesions, polyneuropathy and mononeuropathy.
Musculoskeletal system
Musculoskeletal system involvement with joint deformations is common in SSc, leading to disability with restricted joint mobility. The advanced fibrotic process often leads to joint and tendon entrapping, resulting in pain, contractures or tendon friction rubs [33].
In general, the most common complications of musculoskeletal system in SSc include tendinopathy, joint contractures and in some cases arthritis [34]. In 45% to 90% of SSc cases arthralgia and arthritis of small joints of the hands and wrists and also of the knees and ankles are present. Joint involvement may be a first manifestation of SSc – in its acute or slowly progressive course. Patients often complain of joint stiffness, mainly related to the cutaneous thickening or calcification around the joints. In the majority of cases arthritis is non-erosive and mainly detected in the metacarpophalangeal joints, wrists, knees, distal and proximal interphalangeal joints. Joint pain, markedly more common in dsSSc, is one the most significant symptoms affecting functionality of SSc patients. In addition, joint contractures may cause disability. In about 20% of cases of SSc osteolysis is present [34, 35].
The tendon involvement in SSc most commonly takes the form of tenosynovitis and tendon friction rubs (TFRs), which are also related to rapid skin fibrosis progression [36]. They occur more often at the beginning of the disease and usually in patients with diffuse cutaneous subset, but can be found at all stages and in both subsets of scleroderma. Tendon friction rubs, which can be easily detected by physical examination, are present in various areas, most commonly in tendons of the hands, elbows, knees and ankles. In the case of coexisting pain it is necessary to perform an ultrasound (US) examination and rule out synovitis [37].
The most frequent bone changes in SSc are acroosteolysis (resorption of terminal phalanges) with calcinosis. These changes are associated with digital ischaemia [38]. It may be minimal, involving only one terminal phalanx, or spread – affecting multiple phalanges. Erosive deformation of distal interphalangeal joints can also occur. Additional findings involve intraosseous deposition of calcium. In early stages of SSc arthritic pain may be present with early radiographic changes, similar as in rheumatoid arthritis, although less damaging. Patients with SSc are under increased risk of osteoporosis mainly due to the chronic inflammatory condition, but also because of early menopause, malabsorption or long-term GS therapy.
Myopathy and myositis
Muscle weakness and pain, mainly affecting forearms and hands or proximal muscles of the limbs, are other very frequent symptoms in SSc. Estimates of skeletal myopathy frequency vary approximately from 5 to 96%, since there are no diagnostic consensus criteria [39]. In addition, there is no agreement whether inflammatory myopathy should be considered a disease symptom or simply scleroderma-myositis overlap. Myopathy can be caused by disuse, atrophy or drug side-effects and may be hard to distinguish from the symptoms seen in patients with idiopathic inflammatory myositis. The involvement of limb muscle weakness and limitation of movement can result from myopathy or skin sclerosis, articular changes or tissue sclerosis – the source of those symptoms can be, at times, hard to distinguish.
So far there are no data describing respiratory muscle involvement in SSc, although in cases of SSc-polymyositis/dermatomyositis overlap syndrome respiratory muscles may be affected. Myocarditis in SSc patients was found in association with myositis, confirming that myopathy is an independent risk factor for cardiac involvement (especially left ventricular dysfunction). It is necessary to screen all patients with SSc for muscle involvement in case of suspicion of overlap syndrome. Tests include laboratory testing (autoantibodies and muscle enzymes, e.g. creatine kinase and aldolase), electromyography (EMG), MRI and muscle biopsy [39, 40]. Myopathy in SSc patients still lacks clearly stated criteria. There is still no consensus on whether to consider it a symptom or scleroderma-myositis overlap. So far two patterns of muscle involvement have been identified, taking into consideration manual muscle strength testing, muscle enzyme levels and EMG results. The first one, called “simple myopathy” and considered to be a mild form, is characterized by proximal muscle involvement, normal or slightly increased CK and aldolase and changes in EMG – polyphasic motor unit potentials. In the case of prominent muscle weakness, notably elevated muscle enzymes and certain features in EMG (e.g. positive sharp waves or increased insertional irritability), the “complicated myopathy” pattern can be recognized, representing scleroderma-polymyositis overlapping [41]. Nevertheless, recent studies tend not to support that classification.
Ultrasonography and MRI should be considered as useful tools for detecting synovitis, tendonitis and tenosynovitis in SSc patients [37, 40]. Ultrasound has become a reference tool in imaging of muscles, joints and soft tissues. It is a non-invasive method, enabling multiregional evaluation in just one session. It allows one to detect inflammatory abnormalities concerning joints and also bone structural changes. In 2009 Cuomo et al. [41] investigated 45 patients with SSc and 15 patients with RA. They detected joint and bone changes such as osteophytes, calcinosis, joint space narrowing, joint effusions and synovial proliferation. Furthermore, in the group of SSc patients the frequency of synovitis observed was much higher than that revealed in clinical evaluation, while calcinosis was present exclusively in the SSc group.
The criteria for MRI assessment of muscular involvement in SSc are still undefined, so MRI continues to be an aid in th process of diagnosing myopathy or myositis for now. The new imaging methods that may be helpful in assessing inflammatory myopathies include contrast-enhanced muscle ultrasound, which differentiates inflamed muscles from atrophic ones, and MR techniques, providing information on muscle recruitment, microcirculation and myofibrillar structure. Radiographic findings characterizing joint involvement in SSc include joint space narrowing, erosions, intraarticular calcification, osteoporosis and erosions (although smaller and not as destructive as in RA patients). Electromyography results are abnormal in about 50% of cases at the beginning of the disease and in up to 90% cases at its later stages [42].
Heart involvement
Heart involvement in SSc is a very common phenomenon and the cause of up to 30% of SSc related mortality. Although the cardiovascular system (CVS) is affected both in lcSSc and dSSc, studies show that patients with dSSc suffer from cardiac complications more often, especially when they experience fast progression of skin fibrosis [43].
The frequency of scleroderma heart disease is underestimated due to the fact that it remains asymptomatic for a long time. Probably up to 100% of patients with SSc have a certain degree of cardiovascular involvement, often remaining symptomless. Studies show that only 23–32% of patients with SSc suffer from clinically overt heart complications [44]. Undetected CVS complications in SSc contribute strongly to the rate of sudden death in this group of patients.
The first symptoms of heart involvement usually point to a poor prognosis and are an independent mortality risk factor and account for 20% of deaths in patients with SSc [45]. Therefore SSc heart involvement should be diagnosed as early as possible, during the asymptomatic period in particular. Cardiac disease in SSc may result from primary heart involvement or be secondary to other organ complications, e.g. pulmonary fibrosis. One of the most common cardiopulmonary complications is the development of pulmonary arterial hypertension (PAH) due to interstitial lung disease (ILD) and vasculopathy. Myocardial fibrosis, myositis, coronary heart disease, right and left heart failure, arrhythmias, valvular complications and pericardial disease are manifestations of primary heart involvement. Myocardial fibrosis in SSc has a focal pattern and is distributed equally in the whole myocardium, increasing stiffness and remodeling heart muscle tissue. Therefore it is a potential risk factor of heart failure, even with normal left ventricular ejection fraction (LVEF %).
None of the routinely conducted tests, such as electrocardiography or echocardiography, is specific and sensitive enough for detecting heart fibrosis. Contrast enhanced heart MRI is the only tool to detect scleroderma heart involvement at early stages of the disease – late gadolinium enhancement on MRI imaging is a sign of tissue fibrosis. Cardiac MRI not only detects the fibrosis and myocardial inflammation but also identifies weaker perfusion of heart muscle and potential ventricular dysfunction. Excessive fibrosis, microvascular damage and activation of the immune system result in focal ischemia and myocarditis, which contribute to the damage of the myocardium. Interestingly, studies show that intensification of immunosuppressive therapy in SSc patients with overt cardiac complications leads to reduction of symptoms and normalization of cardiac enzyme concentrations and MRI results – a phenomenon which underlines the role of myocarditis in scleroderma heart disease [44].
There have been many studies conducted on finding a proper serum marker of SSc heart involvement useful in diagnosis at early stages of the disease, with both brain natriuretic peptide (BNP) and N-terminal pro-brain natriuretic peptide (NT-proBNP) being investigate [45]. Both BNP and NT-proBNP are elevated in patients with systemic sclerosis related pulmonary hypertension and may be considered as markers of early pulmonary arterial hypertension. NT-proBNP alone is also a sign of abnormal left and right ventricular contractility, which could also serve as a marker of systemic sclerosis heart involvement. High-sensitivity cardiac troponin T (HS-cTnT) is regarded as a sensitive marker of damage of the myocardium due to inflammation or ischemia, e.g. in acute myocardial infarction or myocarditis. The levels of plasma concentration of HS-cTnT are elevated in disorders with myocardial microvascular damage. Avouac et al. [46] measured plasma concentrations of both NT-proBNP and high-sensitivity cardiac troponin T (HS-cTnT) in patients with SSc, finding that both were elevated. These easily obtained cardiac markers can be used in assessing the cardiovascular risk in patients with systemic sclerosis.
Left ventricular dysfunctions
The process of progressive fibrosis along with myocardial inflammation leads to systolic and diastolic dysfunction of the left ventricle. However, congestive heart failure is a rare condition and is diagnosed at the end stage of scleroderma. Left ventricle contractility dysfunction with abnormal LVEF is rarely observed in scleroderma patients. Left ventricular hypertrophy and impaired diastolic relaxation is a more common cardiac complication of SSc and leads to a reverse mitral inflow and enlargement of the left atrium. Both systolic and diastolic dysfunctions significantly worsen with exercise [44, 47].
The most common method to detect functional heart abnormalities is echocardiography with tissue Doppler imaging. Other methods providing more precise imaging of the dysfunction of the myocardium are MRI, computed tomography (CT) scan and positron emission tomography (PET).
Other complications
Pulmonary arterial hypertension
Pulmonary arterial hypertension and subsequent right heart failure are very important risk factors in patients with SSc. Pulmonary fibrosis and pulmonary vascular damage lead to restriction of blood circulation and result in PAH. Pulmonary arterial hypertension affects about 15% of scleroderma patients and leads to right heart failure [48]. Elevated pressure in pulmonary arteries influences the right ventricle, leading to its insufficiency. The prognosis for SSc patients with pulmonary arterial hypertension, once a fatal complication, has improved due to the early detection and treatment.
The gold standard examination for PAH is right heart catheterization, which shows elevated pressure in the pulmonary artery.
Arrhythmias and conduction defects
Fibrosis of the myocardium and heart conduction system lead to arrhythmias and conduction defects, which are common in scleroderma patients. These abnormalities may be concealed and mild, but they may also be of life-threatening significance. Arrhythmias and conduction defects significantly worsen the prognosis in systemic sclerosis [49]. To reduce the risk of sudden cardiac death, every patient with systemic sclerosis should routinely undergo a standard 12-lead ECG as well as Doppler echocardiography. The next step in diagnosing arrhythmias and conduction defects is 24-hour Holter monitoring and exercise testing.
When the risk of sudden cardiac death is assessed as high, the patient may require implantation of an implantable cardioverter defibrillator (ICD).
Pericardial complications
Involvement of the pericardium in SSc patients is usually asymptomatic. Clinical signs of pericardial complications are seen in 5–16% of patients, while at autopsy the pericardium is affected in up to 72% of patients with SSc [50]. Pericardial involvement may take the form of pericarditis, pericardial effusion, adhesions of pericardium and infrequently tamponade or constrictive pericarditis. Pericardial effusion may also be a result of pulmonary hypertension. Complications such as massive pericardial effusion and pericardial tamponade are rare, but are considered signs of poor outcome [50].
Pulmonary complications
Pulmonary complications of SSc are one of the main causes of morbidity and mortality in the course of SSc. ILD is one of the major types of lung involvement. Most of the patients present cutaneous involvement (Raynaud’s phenomenon, sclerodactyly) before the time when ILD is identified. There is an increased risk of early developing of ILD in diffuse cutaneous systemic sclerosis.
The presence of characteristic autoantibodies – antitopoisomerase I (also known as anti-Scl-70), anti-U3RNP or anti-ThTTo antibodies – supports the diagnosis and increased likelihood of ILD [51]. The vast majority of patients with a lung defect present a pattern termed nonspecific interstitial pneumonia (NSIP). Histopathologically lung disease is characterized by varying degrees of fibrosis and pulmonary inflammation. An early stage of ILD is mostly asymptomatic. At later stages mostly general symptoms such as fatigue, dry cough, breathlessness, and dyspnea develop, and auscultatory crackles or abnormalities on pulmonary function testing (PFTs) or the chest radiograph may be present.
Pulmonary function testing, including forced vital capacity (FVC) and total lung capacity (TLC), as well as a diffusing lung capacity for carbon monoxide (DLCO), is performed widely in SSc patients and is a key in the diagnosis and long-term follow-up. Periodic DLCO estimation is correlated with HRCT findings or 6-minute walking test. Generally, the diagnosis of SSc-associated ILD is based on the inherence of characteristic features of ILD in the high resolution computed tomography (HRCT) of the chest. The NSIP pattern manifests with ground glass opacities in a peripheral distribution with subpleural and basilar predominance. Ground-glass opacities are typically associated with the early stage of the disease. Chest X-ray has inadequate sensitivity, but provides visualization of advanced lesions. The monitoring of FVC, TLC, and DLCO is recommended every 3–6 months.
Renal complications
Systemic sclerosis may also lead to acute or chronic renal complications, of which scleroderma renal crisis (SRC) is the most severe. It occurs in 10% of patients with dSSc and very seldom – 0.5% – in patients with lSSc [52] The pathogenesis of SRC remains unclear. However, recent studies suggest vasculopathy as a source, similarly to other involved organs. An endothelial cell injury may play an important role in SRC, leading to excessive proliferation and thickening of the vessel wall. This process decreases renal blood flow and causes higher renin levels, leading to malignant hypertension. Moreover, previous high dose (> 15 mg daily) prednisone administration has been considered as an independent risk factor for SRC.
The exact pathology is unknown, but a role of corticosteroids in endothelial disturbance has been postulated. There is also a strong association between drugs, which cause vasospasm, such as tacrolimus, cyclosporine or cocaine – and SRC occurrence [52, 53]. There are more predictive factors for SRC: diffuse form of SSc, early disease symptoms (less than 4 years), quick onset and rapid progression of the disease, presence of antibodies against RNA polymerase III, anti- topoisomerase I, anti-U3RNP, anti-Th/To or new cardiac events [52].
However, the kidneys are more frequently affected by a chronic process characterized by gradual decreases in eGFR, increase in serum creatinine concentration, fairly elevated proteinuria and hematuria, and moderate arterial hypertension. Patients mostly present accelerated hypertension with other symptoms associated with it – headache, blurred vision. Very rarely nowadays, in severe cases, symptoms of encephalopathy, such as seizures, may occur. The oliguric renal failure in SSc patients may lead to congestive heart failure, serious arrhythmias resulting even in cardiac arrest or pericardial effusions. Moreover, secondary to malignant hypertension, pulmonary hypertension may occur. Elevated creatinine serum concentration, decrease of glomerular filtration rate (GFR), proteinuria, microangiopathic hemolytic anemia and lower platelet count are typical laboratory findings in renal involvement in SSc. Renal biopsy is usually not required as it does not have much diagnostic value in SSc, apart from instances of diagnosing overlapping syndrome (e.g. with lupus nephritis) or other overlapping renal conditions.
Renal complications result in up to 10% early mortality in SSc, in spite of the improvement in therapy. Scleroderma renal crisis can lead to end stage renal failure in 40–50% of cases, with renal replacement therapy required. This state may be temporary, up to 24 months after the SRC incident. Kidney transplant remains favorable treatment (up to 82% 5-year survival in the French population) for end stage renal failure due to poor outcome of long term dialysis [54].
Prognosis in systemic sclerosis
The prognosis of SSc depends mainly on the severity of internal organ involvement. The presence of manifestations such as cardiomyopathy or severe arrhythmias, advanced lungs fibrosis, and renal failure with SRC risk and advanced skin fibrosis result in a poor prognosis [5, 9, 16]. The prognosis also depends on the patient’s race (a worse course of the disease is observed in the black race) and sex (worse in males). The first three years of the disease are considered as a crucial period for the patient’s prognosis. The progression of the organ involvement is usually faster in this time interval, which proves how important it is to recognize and treat SSc early. The presence of arthritis is associated with vascular and muscular involvement and with an elevated HAQ-DI score (Health Assessment Questionnaire Disability Index). Acro-osteolysis is strongly associated with digital ulcers, calcification, ILD and esophagus involvement. In the case of flexion contractures found in radiographic imaging, the association with ILD, esophagus involvement and increased HAQ-DI score is common.
The presence of TFRs is associated with active disease and high risk of developing renal, cardiac and gastrointestinal complications. In addition, there is strong evidence of associations between TFRs, digital ulcers, muscle weakness, pulmonary fibrosis and renal crisis. Tenosynovitis strongly corresponds with joint space narrowing in the wrist area, demineralization, higher Rodnan skin score and the presence of anti-Scl-70 antibodies. It is more often found in patients with tendon friction rubs and swollen joints. A few autoantibodies are considered closely associated with musculoskeletal involvement in SSc patients, in particular PM/Scl antibody (very often seen in patients with scleroderma and polymyositis overlap) and anti-Ku antibody, mainly associated with muscle weakness, EMG features and CK elevation. Systemic sclerosis patients with PM/Scl and anti-Ku antibodies elevated are more likely to present lSSc rather than dSSc. The likelihood of myositis in SSc is lower in the presence of ACA antibodies and higher in the presence of anti-Scl70. Other, less frequent autoantibodies associated with muscle involvement are anti-U3-RNP antibody (in SSc patients), anti-PL7, anti-PL12, anti-Jo1 and anti-SRP antibodies (in SSc-myositis overlap syndromes).
Changes in the musculoskeletal system may become a cause of the patient’s disability and concern. In an advanced process tendons and joints get entrapped, causing mainly pain and tendon friction rubs. Furthermore, skeletal myopathy and inflammatory arthritis may occur superimposed on the fibrotic process. It is very important to improve activity of involved tissues from the beginning. Muscular involvement in SSc patients does not worsen the prognosis compared to those without myopathy, although the association with a higher risk of myocardial involvement has been confirmed. ILD predicts increased mortality in SSc, as illustrated by a retrospective study: patients with severe ILD had a nine-year survival rate of approximately 30%, whereas patients with SSc who did not have severe involvement of an organ system had a nine-year survival rate of 72% [55].
Pulmonary arterial hypertension with a combination of right heart failure is a well-known poor prognostic factor associated with high mortality risk in SSc. Heart involvement, especially with arrhythmias, conduction defects and pericardial effusions, may lead to sudden cardiac death in this group of patients.
Treatment
The complexity of SSc treatment results from the need to inhibit the autoimmune process, inflammation and organ specific management. As the pathogenesis of SSc is still unclear, the treatment is based on disease-modifying and organ-specific drugs. Therapeutic decisions should be made after appropriate assessment of symptoms, disease duration, activity and complications. Currently, there is no recommended treatment for SSc-associated myopathy. In an inflammatory type of myopathy glucocorticosteroids (GC) are suggested. In case of absence of an inflammatory component, patients often remain untreated [56]. Arthritis in SSc usually requires disease-modifying antirheumatic drugs (DMARDs) or GC. For peripheral vasculopathy and digital ulcers calcium channel blockers (nifedipine, amlodipine) are recommended. In case of a weak response phosphodiesterase type 5 (PDE-5) inhibitors should be introduced. Intravenously administered prostanoids significantly increase the healing process of digital ulcers and improve the microcirculation [6, 56]. In skin fibrosis immunosuppressants, such as methotrexate (MTX), cyclophosphamide (CYC), and mycophenolate mofetil (MMF), are commonly used. When the contraindication or ineffectiveness of therapy occurs, low-dose GCs or rituximab (RTX – anti-CD 20 monoclonal antibody) can be introduced [57].
There were reports in experimental models, as well in studies on humans, that cannabinoids with anti-fibrotic activity may influence dermal fibroblasts and inhibit fibrosis in SSc [58]. The management of GIT manifestations of SSc is based on the symptoms rather than the anatomical site affected. In GERD proton pump inhibitors (PPI) and prokinetic agents are widely administered. To eradicate SIBO, oral antibiotic therapy is used, and rifaximin appears to be the most effective drug [59]. This antibiotic is also proven to be effective in the case of diverticulosis. It is important to implement proper dietary habits to SSc patients to decrease the impact of the symptoms on quality of their life and to avoid the most dangerous complications such as malabsorption and weight loss. In the most severe cases enteral or parenteral feeding supplementation might be beneficial [60]. Patients with SSc who develop musculoskeletal complications should be given individually adjusted immunomodulatory therapy. Methotrexate is most commonly used as first-line treatment, followed by GCs and hydroxychloroquine (HQC). The decision about initiating ILD therapy can be difficult. As patients with SSc-ILD have stable or slowly progressive disease, immunosuppressive therapy is needed in 25–30%. First line therapy is MMF at a dose of 2.0–3.0 g/day p.o.; an alternative to MMF is CYC. Cyclophosphamide is recommended in i.v. pulses for 6–12 months used in ILD with severe, progressive course, unresponsive to MMF treatment. In some cases the agent can be administered via daily oral dosing 1–2 mg/kg per day for 12 months. Intravenous CYC treatments due to the lower cumulative dose (the lifetime cumulative dose of approximately 15 g should not be exceeded) have less frequent adverse effects and the ability to ensure adequate hydration prior to dosing. A therapeutic alternative is RTX in monotherapy or in combination with CYC and autologous stem cell transplantation (ASCT) [61, 62].
There have not been any clinical studies determining the required duration of immunosuppressive therapy in patients with ILD. Experts recommend that therapy should continue for 4–5 years after reaching a stable outcome of pulmonary function tests. Monitoring should be controlled with lung function tests (FVC, TLC, DLCO) every 3–6 months. Pulmonary hypertension in SSc requires therapy with endothelin receptor antagonists such as bosentan or macitentan, phosphodiesterase type 5 (PDE5) inhibitors and an agonist of soluble guanylate cyclase (sGC) such as riociguat [55, 56]. Prostacyclin analogues are also approved for treatment of PAH in SSc. Cyclophosphamide administered in intravenous pulses is recommended in ILD as a first-line therapy with sequential introduction of azathioprine (AZAT) or cyclosporin. Recently a good therapeutic option is MMF. Nintedanib – a tyrosine kinase inhibitor which has antifibrotic and anti-inflammatory properties and is approved in the treatment of idiopathic pulmonary fibrosis (IPF) – has proved to be effective in the treatment of SSc-ILD, but not in SSc with other organ involvement, including skin involvement [63]. The discussion about the usefulness of cannabinoids, with their anti-fibrotic and anti-inflammatory properties, in the treatment of autoimmune diseases is still ongoing. Currently, clinical trials with cannabinoids are under way, with positive effects on skin reported. Also, the future of cannabinoids in the treatment of ILD in SSc is being considered [58].
The basic SSc treatment includes adequate control of systemic hypertension. Introducing angiotensin-converting-enzyme inhibitors (ACE-I) played a significant role in SRC outcomes. Early diagnosis of SRC and administration of ACE-I may prevent serious complications. ACE-I reduces angiotensin levels, despite higher concentration of renin. ACE-I also cause higher levels of bradykinin, which is a well-known vasodilator. Angiotensin receptor blockers (ARB) do not effect bradykinin levels. This may explain why ARB are not so beneficial in SRC as ACE-I, although the process is not yet fully understood. In cases of normotensive SRC a low doses of ACE-I may be used. Also other hypotensive agents may be used to control hypertension (calcium blockers, nitrates, ARB) along with ACE-I. The cardiac function must be monitored closely as anti-hypertensive drugs may cause relative hypovolemia. Beta-blockers are not recommended due to their worsening effect on Raynaud’s phenomenon and vasoconstriction. Recent case reports show potential beneficial effects of direct renin inhibitors and bosentan, a selective endothelin A receptor antagonist. Nevertheless, further studies are required to evaluate their effectiveness in SRC.
In the discussion of SSc treatment, hematopoietic autologous stem cell transplantation (HASCT) although still being developed and discussed, confirmed its effectiveness in SSc in the ASTIS [61] and ASSIST [62] studies, which confirmed improvement in mRSS and concluded that the main target population for HASCT is the group of patients with early diffuse SSc. They also highlighted the importance of proper patient selection for HASCT and of the post-transplant management. The mortality rate of ASCT depends on the total dose of CYC and a more aggressive myeloablative conditioning method [62]. The ASTIS trial demonstrated that in the HASCT group treatment-related mortality was increased in the first year after the transplant, while in the next years there was a significant positive effect (long-term event-free survival) of HASCT compared to the control group [61]. Current treatment of SSc and further perspectives are summarized in Table II.
Table II
Current systemic sclerosis treatment and further perspectives
Conclusions
Systemic sclerosis is still undoubtedly a challenge for clinicians, because of the variety of symptoms. SSc course and prognosis depend on clinical picture and character of organ involvement (kidney, heart and lungs in particular). The treatment of SSc should be carried out by rheumatologists consulting other specialists. Although early diagnosis and new therapeutic options significantly improve SSc prognosis, it is still characterized by a severe course and high risk of early death.