
Abstract
Longitudinal piezoelectric constant (e33) values of wurtzite materials, which are listed in a structure database, are calculated and analyzed by using first-principles and statistical learning methods. It is theoretically shown that wurtzite materials with high e33 generally have small lattice constant ratios (c/a) almost independent of constituent elements, and approximately expressed as e33 ∝ c/a − (c/a)0 with ideal lattice constant ratio (c/a)0. This relation also holds for highly-piezoelectric ternary materials such as ScxAl1−xN. We conducted a search for high-piezoelectric wurtzite materials by identifying materials with smaller c/a values. It is proposed that the piezoelectricity of ZnO can be significantly enhanced by substitutions of Zn with Ca.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
In recent times, owing to their suitable mechanical and semiconducting properties, piezoelectric wurtzite semiconductors such as ZnO and GaN have received a lot of attention in the form of piezotronics and piezo-phototronics device materials.1) The wurtzite-type piezoelectric materials, especially AlN, have another advantage that these can be used in high-temperature environments, e.g., as sensors in automobile engines, because their noncentrosymmetric structures are stable even at high temperatures.2,3) However, the piezoelectric constants of wurtzite-type materials are generally much smaller than those of the perovskite-based materials such as Pb(ZrxTi1−x)O3, known as PZT, by few orders. It remains a challenge to explore better piezoelectric wurtzite materials; there are many reports aiming to enhance piezoelectricity by element doping or codoping into parent materials.4–7)
Among the wurtzite materials, the highest piezoelectricity has been experimentally discovered for ScxAl1−xN (about 25 pC/N for x ∼ 0.5).2,3) First-principles calculations have shown the microscopic effects of Sc on the piezoelectricity in ScxAl1−xN by demonstrating an enhancement in piezoelectricity with increase in x.8,9) Tasnádi et al. have revealed that the piezoelectric enhancement comes from the large responses of atomic positions to external strain and the significant softening of elastic constant.8) However, novel materials which are superior to ScxAl1−xN have not been synthesized yet as there are no clear and general material-design criteria practically usable for enhancing the piezoelectricity of wurtzite materials. It has been proposed theoretically that there exists a relationship between piezoelectricity and microscopic structures,10) but the physical mechanism behind the relation with intricate parameters is not fully understood yet. Therefore, better understanding of piezoelectricity in wurtzite materials might lead to simple design criteria making it possible to find a controllable key parameter describing wurtzite piezoelectricity with the help of first-principles and machine learning techniques which have generated a great interest in the recent times.11,12)
In this study, we calculate longitudinal piezoelectric constants (e33) by first-principles method for more than a dozen binary wurtzite materials and investigate the relationships between the piezoelectric constants and several material parameters using the statistical learning methods. We show that a strong correlation exists between e33 and the lattice parameter ratio (c/a), providing a guiding principle that higher piezoelectricity can be obtained for smaller c/a ratio. This guiding principle could be applied not only for binary materials but also for ternary materials of doped systems. Based on the above relation, we have explored highly piezoelectric wurtzite materials by uncovering small c/a materials.
The wurtzite crystal structure has three independent structure parameters, i.e., the lattice constants (a and c) and the internal atomic coordinate along the c-axis (u). In our study we calculated the parameters for all the binary wurtzite crystals listed in the structure databases. a, c, and u are fully optimized from the first principles for all the calculations. The first-principles calculations are based on the density functional theory (DFT) within the Perdew–Burke–Ernzerhof generalized gradient approximation (GGA) to the exchange and correlation functionals.13,14) The computations were performed using the HiLAPW code, which employs the all-electron full-potential linearized augmented plane wave (FLAPW) method.15–17) Longitudinal piezoelectric constant along c-axis (e33) was calculated by means of the Berry phase method.18–20) Statistical data analyses were performed using the statistical computing software R with the glmnet-package; this package fits the least absolute shrinkage and selection operator (LASSO) and the elastic-net regularized generalized linear model (GLM) for regression.21,22) The GLM analysis can identify meaningful variables describing a target variable among a given set of explanatory variables.
Table I shows the calculated equilibrium structure parameters (a, c, c/a, and u), piezoelectric constants (e33), Born effective charges  , band gaps (Eg), and elastic constants (C33) of eighteen binary wurtzite materials which are listed in the Inorganic Crystal Structure Database (ICSD).23) The calculated lattice parameters are in good agreement with the typical experimental values listed in ICSD less than 4% error, except for BP. The calculated values of a and c of BP underestimate the ICSD data (only one data listed as ICSD Code 615155) by 10%. Experimental values of e33 are available only for a limited number of materials, for example, 1.55 C/m2 for AlN and 0.96 C/m2 for ZnO; the calculated values of e33 are consistent with the available experimental values and also with the previous calculations.24–28) Some materials such as BN show negative values of e33 as previously reported;26) the negative piezoelectricity has recently attracted much attention.29) Among the calculated binary materials, AlN and ZnO are known to exhibit better piezoelectric performances.
, band gaps (Eg), and elastic constants (C33) of eighteen binary wurtzite materials which are listed in the Inorganic Crystal Structure Database (ICSD).23) The calculated lattice parameters are in good agreement with the typical experimental values listed in ICSD less than 4% error, except for BP. The calculated values of a and c of BP underestimate the ICSD data (only one data listed as ICSD Code 615155) by 10%. Experimental values of e33 are available only for a limited number of materials, for example, 1.55 C/m2 for AlN and 0.96 C/m2 for ZnO; the calculated values of e33 are consistent with the available experimental values and also with the previous calculations.24–28) Some materials such as BN show negative values of e33 as previously reported;26) the negative piezoelectricity has recently attracted much attention.29) Among the calculated binary materials, AlN and ZnO are known to exhibit better piezoelectric performances.
Table I. Calculated lattice constants [a and c (Å)], their ratios (c/a), internal atomic coordinates (u), piezoelectric constants [e33 (C/m2)], Born effective charges  , band gaps [Eg (eV)], and elastic constants [C33 (GPa)] of binary materials with the wurtzite structure. (Note that wurtzite-MgO is a hypothetical material, which is not listed in ICSD. e33 of InN is calculated excluding the Γ point.)
, band gaps [Eg (eV)], and elastic constants [C33 (GPa)] of binary materials with the wurtzite structure. (Note that wurtzite-MgO is a hypothetical material, which is not listed in ICSD. e33 of InN is calculated excluding the Γ point.)
| a | c | c/a | u | e33 | 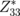 |
Eg | C33 | |
|---|---|---|---|---|---|---|---|---|
| MgO | 3.32 | 5.12 | 1.546 | 0.392 | 1.70 | 1.89 | 3.43 | 164 |
| AlN | 3.13 | 5.02 | 1.603 | 0.382 | 1.42 | 2.69 | 3.98 | 388 |
| ZnO | 3.29 | 5.30 | 1.611 | 0.380 | 1.04 | 2.17 | 0.82 | 185 |
| InN | 3.58 | 5.78 | 1.615 | 0.379 | 0.94 | 3.07 | 0 | 215 |
| GaN | 3.23 | 5.27 | 1.630 | 0.377 | 0.44 | 2.76 | 1.52 | 290 |
| SiC | 3.10 | 5.08 | 1.641 | 0.376 | 0.33 | 2.82 | 2.57 | 501 |
| BP | 3.20 | 5.31 | 1.658 | 0.374 | 0.32 | 0.46 | 1.20 | 525 |
| CdS | 4.20 | 6.82 | 1.623 | 0.377 | 0.31 | 2.26 | 1.13 | 87 |
| CuH | 2.87 | 4.56 | 1.591 | 0.382 | 0.22 | 0.55 | 0.62 | 144 |
| BeO | 2.72 | 4.41 | 1.624 | 0.378 | 0.21 | 1.84 | 7.48 | 485 |
| MgTe | 4.61 | 7.49 | 1.626 | 0.377 | 0.18 | 2.00 | 2.37 | 58 |
| CdSe | 4.39 | 7.14 | 1.626 | 0.377 | 0.15 | 2.36 | 0.55 | 69 |
| ZnS | 3.85 | 6.30 | 1.638 | 0.375 | 0.09 | 2.06 | 2.11 | 144 |
| AgI | 4.68 | 7.70 | 1.645 | 0.373 | 0.05 | 1.30 | 1.38 | 41 |
| LiI | 4.56 | 7.41 | 1.624 | 0.376 | 0.04 | 1.07 | 4.39 | 22 |
| CdTe | 4.68 | 7.67 | 1.641 | 0.374 | −0.04 | 2.33 | 0.61 | 56 |
| CuI | 4.30 | 7.07 | 1.644 | 0.374 | −0.04 | 1.12 | 1.19 | 71 |
| ZnSe | 4.05 | 6.64 | 1.642 | 0.374 | −0.10 | 2.15 | 1.21 | 103 |
| BN | 2.56 | 4.24 | 1.653 | 0.375 | −1.08 | 1.94 | 5.59 | 1112 |
From Table I, we can roughly observe a general trend that the value of e33 increases with decreasing value of c/a indicating that the structural parameter could be a key index describing the piezoelectricity of wurtzite materials. Value of u also increases with decreasing c/a, showing a strong correlation between the structure parameters. The calculation result shows that e33 does not clearly correlate with the values of the electronic structure parameter Eg and energy profile parameter C33. Note that Eg calculated using DFT-GGA usually underestimates the experimental values. To confirm the above trend, we performed calculations on a hypothetical material MgO with wurtzite structure, which can have much smaller c/a.30) The calculated values of lattice parameters are consistent with the previous result for wurtzite MgO.30) Calculated value of e33 and c/a of wurtzite MgO support the relation of higher e33 for smaller c/a. Note that the thermodynamically stable structure of MgO is cubic rock-salt type. The structure stabilities of wurtzite MgO have been theoretically studied and it has been concluded that the wurtzite phase transforms to the nonpolar hexagonal structure as an energetically more stable phase.30,31)
Figure 1(a) shows e33 versus c/a of the calculated wurtzite materials. We can see that e33 tends to increase with decreasing c/a, although most of the materials for which the calculations have been performed are concentrated in a narrow range of e33 less than ±0.5 C/m2 and c/a close to the ideal value of  . In order to know where the doped materials fall in this graph, we plotted the calculated values of e33 for four Sc0.5Al0.5N models.9) Interestingly, the ternary Sc0.5Al0.5N values are seen as an extension of the result of binary materials, showing that there exists a roughly linear relation between e33 and c/a as e33 ∝ c/a (we omit intercept terms in the main text). Figure 1(b) shows the calculated values of u versus c/a for the wurtzite materials. We clearly see that there exists a strong linear correlation as u ∝ c/a. Therefore, c/a or u can be the most important key parameters describing e33.
. In order to know where the doped materials fall in this graph, we plotted the calculated values of e33 for four Sc0.5Al0.5N models.9) Interestingly, the ternary Sc0.5Al0.5N values are seen as an extension of the result of binary materials, showing that there exists a roughly linear relation between e33 and c/a as e33 ∝ c/a (we omit intercept terms in the main text). Figure 1(b) shows the calculated values of u versus c/a for the wurtzite materials. We clearly see that there exists a strong linear correlation as u ∝ c/a. Therefore, c/a or u can be the most important key parameters describing e33.

Download figure:
Standard image High-resolution image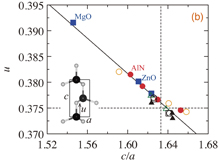
Fig. 1. (a) Piezoelectric constants (e33) and (b) internal atomic positions (u) versus lattice constant ratios (c/a) calculated for wurtzite materials. Calculated binary materials listed in Table I are shown by red closed circles (nitrides), blue closed squares (oxides), green open triangles (sulfides), black open circles (selenides), black open squares (tellurides), black closed triangles (iodides), and yellow open circles (others). In the upper panel, red open circles show calculated values of Sc0.5Al0.5N models.9) Linear fitting lines are shown by solid lines, and the c/a and u values of the ideal wurtzite structure are shown by broken lines.
Download figure:
Standard image High-resolution imageTo further learn about the calculated e33 data statistically, we performed the GLM analyses on these data in which the K-fold cross validations (here we set K = 6) were performed to obtain the best penalization parameters giving a minimum error between GLM and DFT results. We set 56 explanatory variables (descriptors) which included materials data (Eg and C33), structure data (a, c, volume V, c/a, and u), atomic data of constituent elements [anion's and cation's atomic numbers (Na and Nc), nominal ionic valences (Zv), ionic radii (Ra and Rc),32) and electronegativities (χa and χc)33)], and many combinations of arithmetic operations among them. The target variable is set to be e33, and only five descriptors of c/a, (χa − χc)/χaχc, (χa − χc)/(χa + χc), (χa − χc)2, and (Na − Nc)−1 are selected to be nonzero terms. The result shows that c/a is the most dominant descriptor, and others have negligibly tiny contributions with much smaller coefficients than that of c/a (by one or two orders). Therefore, the GLM analysis also shows the relationship of e33 ∝ c/a.
We should note that, when e33/Zv is chosen to be a target variable for the GLM analysis, the minimum error between GLM and DFT results can be slightly reduced compared to that of the target variable of e33. With e33/Zv as the target variable, the selected nonzero descriptors are c/a, (Ra − Rc)−1, (χa − χc)2, and (Na − Nc)−1. In this case, c/a is the dominant descriptor and others have coefficients much smaller (by about two orders of magnitude) than that of c/a; this is roughly described as e33/Zv ∝ c/a. This result is quite natural considering the definition of piezoelectric constants, i.e., higher ionic valences can contribute to higher piezoelectric constants of materials. It should also be noted that the descriptors including the electronegativity differences (χa − χc) are always selected by the GLM analyses, though they have only minor contributions. It has been reported that the value of c/a of wurtzite materials strongly correlates with the square of electronegativity difference,34) indicating that a smaller value of c/a can be obtained for wurtzite materials with larger (χa − χc). It has also been pointed out that a larger e33 might be obtained for materials with larger (χa − χc).3) Recent research shows that the relation between piezoelectricity and c/a holds for wurtzite materials, such as codoped AlN films.35) To the best of our knowledge, it has been not theoretically clarified until now that e33 correlates strongly with c/a.
Here we discuss the microscopic mechanism behind the relation of e33 ∝ c/a [or the better description of e33 ∝ Zv(c/a)]. By the definition of P3 = e33ε3, the piezoelectric constants e33 can be calculated by the first principles as
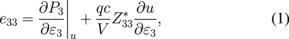
where P3 and ε3 are the electric polarization and the external strain along the c-axis, respectively, and q is the electron charge.9,20,25) For the calculated wurtzite materials, the first (clamped ion) term shows relatively small negative values and does not significantly depend on material. The second (internal strain) term dominantly contributes to the trend of e33 versus c/a shown in Fig. 1(a); thus, Eq. (1) can be approximately expressed as  .
.  can be roughly approximated with Zv, and now we need to understand how ∂u/∂ε3 behaves in wurtzite materials.
can be roughly approximated with Zv, and now we need to understand how ∂u/∂ε3 behaves in wurtzite materials.
Figure 2 shows the calculated total energies E, u, and u' = ∂u/∂(c/a) as a function of c/a for GaN, AlN, ZnO, and MgO with wurtzite structures. We calculate these values by changing c/a via optimization of u; the volume is fixed at each equilibrium value. GaN, AlN, and ZnO have the energy minimum at each c/a listed in Table I in their wurtzite phases, while the energy minimum of MgO is found to be in the nonpolar hexagonal phase with c/a ∼ 1.2 and u = 0.5; it has a flattened layered structure with an inversion symmetry [see the inset in Fig. 2(b)]. For all of the materials, we can see that u gradually increases as c/a decreases, reaching a value u = 0.5 at c/a smaller than about 1.2, almost independent of materials. u', which is the slope of the u versus c/a curve, decreases as c/a decreases from 1.9 reaching the minimum u' values at c/a ∼ 1.2–1.3, after which it quickly diminishes to zero as c/a becomes further smaller. In the c/a range higher than about 1.5 [the c/a range shown in Fig. 1(a)], u' is roughly proportional to c/a. Therefore, in this c/a range, we can approximate as ∂u/∂ε3 ∝ c/a, indicating that Eq. (1) can be approximated as

This rough analysis explains our result of the GLM computation.
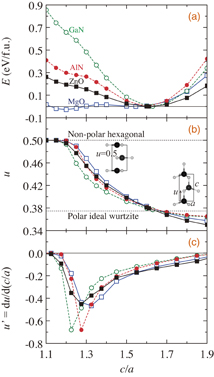
Fig. 2. (a) Total energies (E), (b) internal atomic positions (u), and (c) slopes of u (u') as a function of lattice constant ratios (c/a) for GaN (green open circles), AlN (red closed circles), ZnO (black closed squares), and MgO (blue open squares) with wurtzite structures. Cell volumes are fixed to each equilibrium value.
Download figure:
Standard image High-resolution imageFigure 2(c) implies that the highest value of e33 can be realized for materials with c/a ∼ 1.2–1.4 (near the minimum value of u' depending on materials) if materials can exist stably in the wurtzite phases. It is also expected that piezoelectricity rapidly diminishes and disappears for materials with smaller c/a values which are less than the minimum u' points, because structures in such cases transform to the nonpolar hexagonal phases. Near the phase boundaries between the polar wurtzite and the nonpolar hexagonal structures, e33 would exhibit significant nonlinear changes as Fig. 2(c), thus giant piezoelectricity could be obtained beyond the relation of e33 ∝ (c/a). The result shows that the highest piezoelectricity in wurtzite materials would be generally obtained at c/a near the phase boundary between the polar wurtzite and the nonpolar hexagonal structures, similar to the ScxAl1−xN case.8)
Based on the insight above, we have tried to search for novel highly-piezoelectric wurtzite materials by finding materials possessing smaller c/a values. The most practically useful way for enhancing e33 is element-doping into stable wurtzite materials, as previously conducted. For our study, we chose ZnO as the base material. There have been many reports on the effects of element doping into ZnO, and it has been reported that c/a of ZnO is significantly reduced by Ca doping,37) compared to other elements. A 16-atom super cell model of ZnO was used to calculate the value of e33 of the doped systems by substituting Zn with alkaline earth metals. We considered all the possible combinations of substitution sites and the results were averaged over the models in the same manner as in the previous calculations of ScxAl1−xN.9) Figure 3(a) shows plots of c/a and e33 for A0.5Zn0.5O (A = Mg, Ca, Sr, and Ba). A strong correlation of higher e33 for smaller c/a even in ternary systems can be seen. The smallest c/a and the highest e33 were calculated for A = Ca. For A = Ca, we calculated CaxZn1−xO models in order to optimize the value of x for achieving the highest e33. Figure 3(b) shows c/a and e33 of CaxZn1−xO (x = 0, 0.25, 0.5, 0.75, and 1). We predict that the highest e33 is obtained around x = 0.5. For x higher than 0.5, c/a approaches to about 1.2 — a value at which the atomic structures show almost the nonpolar hexagonal structures having u ∼ 0.5 [see the representative model structures of x = 0.25 and 0.75 shown in the inset in Fig. 3(b)]. There is another practical reason for piezoelectricity reduction at a higher x; non piezoelectric rock-salt structures become more stable compared to the wurtzite phases for higher values of x,36,37) e.g., for x higher than about 0.3 in CaxZn1−xO.37) Therefore, if the wurtzite structure is retained, CaxZn1−xO with x ∼ 0.5 could give the best piezoelectric performance among the materials for which the calculations were performed. Though the calculated value of e33 of CaxZn1−xO is smaller than that of ScxAl1−xN, we expect that CaxZn1−xO has an advantage concerning materials costs and earth abundances of constituent elements.

Download figure:
Standard image High-resolution image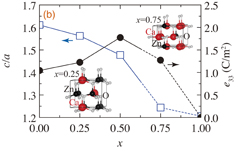
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 3. (a) Lattice constant ratios [c/a (blue squares with left scale)] and piezoelectric constants [e33 (black circles with right scale)] of the A0.5Zn0.5O (A = Mg, Ca, Sr, and Ba) models with wurtzite structures. (b) Calculated c/a (blue squares with left scale) and e33 (black circles with right scale) of the CaxZn1−xO models with wurtzite structures as a function of x. The insets show the representative optimized structures for x = 0.25 and 0.75 models. Values are averaged over calculated models as described in the main text.
Download figure:
Standard image High-resolution image{kind=link}
In conclusion, we have proposed a possible strategy for maximizing piezoelectricity of wurtzite materials. From the first principles calculations with the help of the statistical learning methods, we showed that e33 linearly correlates with c/a (in the ranges higher than about 1.3). Based on the relation e33 ∝ c/a − (c/a)0 [or the better description of e33 ∝ Zv{c/a − (c/a)0}], we have demonstrated some examples for enhancing e33 of materials in wurtzite phases. We have shown that the piezoelectricity of ZnO can be largely enhanced by substitution of Zn with Ca.
Acknowledgments
We would like to thank Akihiko Teshigahara for helpful discussions. This work was supported by JSPS KAKENHI Grant Numbers JP17K04983 and JP16H02267. The computation in this work has been partly done using the facilities of the Supercomputer Center, Institute for Solid State Physics, University of Tokyo.