
The role of 2,4-dihydroxyquinoline (DHQ) in Pseudomonas aeruginosa pathogenicity
- Published
- Accepted
- Received
- Academic Editor
- Timothy Read
- Subject Areas
- Microbiology, Infectious Diseases
- Keywords
- Pseudomonas aeruginosa , Quorum sensing, Pathogenicity, Cystic fibrosis, Pseudomonas quinolone signal, Transcriptional regulation
- Copyright
- © 2016 Gruber et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2016. The role of 2,4-dihydroxyquinoline (DHQ) in Pseudomonas aeruginosa pathogenicity. PeerJ 4:e1495 https://doi.org/10.7717/peerj.1495
Abstract
Bacteria synchronize group behaviors using quorum sensing, which is advantageous during an infection to thwart immune cell attack and resist deleterious changes in the environment. In Pseudomonas aeruginosa, the Pseudomonas quinolone signal (Pqs) quorum-sensing system is an important component of an interconnected intercellular communication network. Two alkylquinolones, 2-heptyl-4-quinolone (HHQ) and 2-heptyl-3-hydroxy-4-quinolone (PQS), activate transcriptional regulator PqsR to promote the production of quinolone signals and virulence factors. Our work focused on the most abundant quinolone produced from the Pqs system, 2,4-dihydroxyquinoline (DHQ), which was shown previously to sustain pyocyanin production and antifungal activity of P. aeruginosa. However, little is known about how DHQ affects P. aeruginosa pathogenicity. Using C. elegans as a model for P. aeruginosa infection, we found pqs mutants only able to produce DHQ maintained virulence towards the nematodes similar to wild-type. In addition, DHQ-only producing mutants displayed increased colonization of C. elegans and virulence factor production compared to a quinolone-null strain. DHQ also bound to PqsR and activated the transcription of pqs operon. More importantly, high extracellular concentration of DHQ was maintained in both aerobic and anaerobic growth. High levels of DHQ were also detected in the sputum samples of cystic fibrosis patients. Taken together, our findings suggest DHQ may play an important role in sustaining P. aeruginosa pathogenicity under oxygen-limiting conditions.
Introduction
Pseudomonas aeruginosa possesses a versatile metabolic system, which facilitates fitness of the organism to survive in diverse environments. People frequently come in contact with P. aeruginosa, but an intact immune system can block the bacterium from establishing an infection. However, both immunocompetent and immunocompromised individuals are still at risk of P. aeruginosa infections, particularly those with diabetes, burn wounds, an implanted medical device, or use contaminated contact lenses (Chatterjee et al., 2014; Gaynes & Edwards, 2005; Gordon et al., 2001). Patients with the genetic disorder cystic fibrosis (CF) are especially susceptible to P. aeruginosa lung infection, which is correlated with increased morbidity and mortality (Hoiby, 2011). In the lungs of a CF patient, P. aeruginosa replicates to high densities and forms antibiotic-resistant biofilms, which is a community lifestyle protected by exopolymeric substance (Palmer et al., 2005). Treatment of chronic and acute P. aeruginosa infections is difficult due to various intrinsic antimicrobial resistance mechanisms, including cell envelope barrier, efflux pumps, and high mutation rate (Henrichfreise et al., 2007; Mandsberg et al., 2009). Compared with classical antibiotics that target essential cellular processes for survival, an alternative approach is to target cellular systems that are responsible for virulence. This strategy may allow the host’s immune system to naturally combat the infection, while not imposing selective pressure for resistant mutants.
Quorum sensing (QS) is a form of bacterial intercellular communication that synchronizes group behaviors and is essential for P. aeruginosa to establish an infection (Rumbaugh et al., 1999). QS molecules bind to their cognate transcriptional regulators at a critical intracellular concentration to promote virulence factor production. The Las and Rhl systems, commonly found in gram-negative bacteria, produce acylhomoserine lactones (AHLs) that activate LuxR-type transcriptional regulators LasR and RhlR (Fuqua & Greenberg, 2002). The Pqs system, which is limited to P. aeruginosa and other closely related bacteria, produces alkylquinolones that activate a LysR-type transcriptional regulator PqsR in P. aeruginosa (Diggle et al., 2006). Among the greater than 50 different quinolones produced by the Pqs system, only alkylquinolones HHQ and PQS have established functions for signaling and promoting pathogenicity, such as activating the production of pyocyanin and biofilm formation (Deziel et al., 2004).
Alkylquinolones are synthesized by enzymes encoded by the pqs operon and other pqs genes located elsewhere on the genome. PqsA catalyzes the first step in quinolone synthesis by converting anthranilic acid, an intermediate of tryptophan biosynthesis, to anthraniloyl-CoA (Coleman et al., 2008). The next step involves the transfer of the anthraniloyl-moiety to PqsD and the release of CoA (Bera et al., 2009). PqsD condenses the anthraniloyl moiety with malonyl-CoA to form 2,4-Dihydroxyquinoline (DHQ), the only terminate non-alkylated quinoline species (Zhang et al., 2008). Formation of 2-heptyl-4-quinolone (HHQ) requires PqsD, PqsB, and PqsC to condense longer chain fatty acids with anthraniloyl-CoA (Bredenbruch et al., 2005). PqsH converts HHQ to 2-heptyl-3-hydroxy-4-quinolone (PQS) in the presence of oxygen (Schertzer, Brown & Whiteley, 2010). Unsaturations, different alkyl chain lengths, and modification of the ring-substituted nitrogen generate the diversity of alkylquinolones (Deziel et al., 2004). Together, the diversity of quinolones and their effects on P. aeruginosa pathogenicity is still not well understood.
In addition to its functions in cell-to-cell communication, PQS exhibits other biological activities that are also important in P. aeruginosa virulence (Dubern & Diggle, 2008; Williams & Camara, 2009). However, in hypoxic environments such as the thick mucus of the CF lungs, PQS production ceases due to the lack of oxygen (Schobert & Jahn, 2010; Su & Hassett, 2012). In contrast, DHQ is the most abundant quinolone in P. aeruginosa planktonic cultures and, unlike PQS, its synthesis does not require oxygen (Zhang et al., 2008). Our previous work showed that mutants only producing DHQ maintained pyocyanin production and antifungal activity of P. aeruginosa (Rella et al., 2012). However, the role of DHQ is not well understood and its determination may further help understand the regulatory activities within the Pqs system and identify new strategies for anti-Pseudomonas treatments. In this study, we investigated the mechanism by which DHQ affects P. aeruginosa virulence, regulates pqs operon transcription through PqsR, and interacts with PqsR. To mimic the environment colonized by P. aeruginosa during chronic lung infections, we also determined the effect on quinolone composition during growth with different amounts of available oxygen and nutrients. Finally, we assessed CF patient sputum samples for quorum-sensing molecules by mass spectrometry analysis. Our findings suggest that DHQ plays a role in P. aeruginosa pathogenicity and may help to establish P. aeruginosa infection under oxygen-limiting conditions.
Methods
Bacterial strains, plasmids, and media
P. aeruginosa wild-type strain PAO1 and its derived mutants, and E. coli strains were grown in Luria–Bertani (LB) medium at 37 °C in a shaker incubator. Cystic fibrosis mimic media was prepared according to Palmer et al. (2005). Hypoxic conditions were generated by flushing media with N2 for 10 min and incubating cultures in a screw-cap vial sealed with a Tephlon© insert and nitrogen-filled head space. Antibiotic concentrations for bacterial selection were: 50 µg/ml and 200 µg/ml carbenicillin (CBC) for E. coli and P. aeruginosa, respectively, 30 µg/ml kanamycin (Kan), 34 µg/ml chloramphenicol (Cam), 30 µg/ml gentamicin (Gm). Culture density was assessed by measuring absorbance at 600 nm. DHQ (2,4-dihydroxyquinoline) and farnesol were purchased from Sigma-Aldrich and PQS (2-heptyl-3-hydroxy-4-quinolone) and HHQ (2-heptly-4-quinolone) were purchased from Qingdao Vochem Co.
Generation of mutants
P. aeruginosa mutant strains ΔpqsAB, ΔpqsB, and ΔpqsC were generated by homologous recombination using a protocol described previously (Choi & Schweizer, 2005). The mutant alleles were constructed by overlapping PCR to contain a gentamicin-resistance cassette flanked by 5′ and 3′ fragments of the gene to be deleted. The mutant fragments were inserted into pEX18ApGW, a suicide vector, to produce gene knockout plasmids. Each knockout plasmid was transformed into E. coli strain SM10 and conjugally transferred from SM10 to PAO1. The resultant integrants were selected on PIA medium containing 30 µg/ml gentamicin (Gm30). To resolve merodiploids, Gm-resistant colonies were streaked for single colonies on LB+Gm30 plates containing 5% sucrose. The unmarked deletion mutants were generated by Flp-mediated marker excision utilizing pFLP2. Potential mutants were screened by PCR using corresponding flanking primers and were confirmed by sequencing.
Generation of protein-expressing plasmids and protein purification
Coding sequences of PqsR and PqsR-C87, a truncated version of PqsR containing the ligand-binding domain starting from Cys87, were amplified by PCR and cloned into a modified pET-28a vector to express recombinant PqsR and PqsR-C87 as His-tagged SUMO fusion proteins for increased solubility. E. coli strain BL21(DE3) or Rosetta containing the expression plasmids was grown in LB with antibiotics (Kan for BL21(DE3), Kan and Cam for Rosetta) to OD600 of 0.6 and heat-shocked at 45 °C for 45 min. Overexpression of recombinant protein was induced by 0.1 mM IPTG at 16 °C for 16 h. The cells were harvested by centrifugation, resuspended in 20 mM Tris–HCl (pH 8.0) with 500 mM NaCl, and lysed by sonication. His-tagged protein in the cell-free extract was purified by nickel affinity chromatography. Fractions containing pure protein were pooled, concentrated, and stored at −80 °C. Protein concentration was determined using the Bradford method with γ-globulin as the protein standard.
Caenorhabditis elegans survival and imaging
Bristol N2 strain of C. elegans was maintained and synchronized on nematode growth media (NGM) containing E. coli OP50 according to the WormBook (Girard et al., 2007). Synchronized L4 nematodes (around 30 per plate) were transferred to 60 mm petri dishes containing a lawn of the tested P. aeruginosa strain. The antimetabolite 5-fluorouracil (25 µM) was added to the NGM agar to inhibit worm reproduction during the course of the experiment. The plates were incubated at 25 °C and worms were monitored daily for survival. Worms were determined dead when no movement was observed following stimulation with a platinum wire. To assess in vivo bacterial colonization, P. aeruginosa strains PAO1, ΔpqsAB, and ΔpqsB carrying the GFP-expressing plasmid pSMC2 (Bloemberg et al., 1997) were used for infection. At the indicated time-points, nematodes were transferred onto fresh agar to remove free-living bacteria, placed into 10 µl of water on a glass slide, and viewed using a Nikon TE2000-S Epifluorescent microscope with a CRI-Nuance imaging system. Images were color-enhanced and analyzed with the ImageJ software.
Pyocyanin quantification
Pyocyanin concentration was measured using a modified protocol from previous work (Das & Manefield, 2012). Briefly, 400 µl of cell-free supernatant from an overnight culture was mixed vigorously with 240 µl chloroform. After organic-phase separation, 200 µl of the organic phase was transferred to a new tube and mixed with 60 µl 0.2 N HCl. Pyocyanin was measured spectrophotometrically at 520 nm using a NanoDrop ND-1000 spectrophotometer.
Quantitative real-time PCR (qRT-PCR)
P. aeruginosa strains were grown in LB until early stationary phase. Total RNA was extracted, treated with DNaseI, and precipitated overnight at −20 °C. cDNA was generated using the Bioline Tetro cDNA synthesis kit. Real-time quantitative PCR was performed on a Bio-Rad MyiQ single-color real-time PCR detection system. The primers used for each gene were as follows: for pqsA, forward primer (TTCTGTTCCGCCTCGATTTC) and reverse primer (AGTCGTTCAACGCCAGCAC); for rpoD, forward primer (GGGCGAAGAAGGAAATGGTC) and reverse primer (CAGGTGGCGTAGGTGGAGAAC). PCR reactions were performed in a 96-well plate with a reaction mixture containing 10 µl SensiFAST SYBR Green mix (Bioline Reagents, London, UK), 200 ng cDNA, and 200 nM each of forward and reverse primers in a total volume of 20 µl. All reactions were performed in triplicate. The thermal cycling conditions were set at 95 °C for 3 min, followed by 40 cycles of 2-step amplification (10 s at 95 °C and 45 s at 57 °C). Data were analyzed with MyiQ software. The abundance of pqsA gene was normalized against the house-keeping rpoD gene using the ΔΔCt method.
pqsA’-LacZ fusion reporter assay
LacZ reporter assay was performed in both E. coli and in P. aeruginosa strains as previously described (Cugini et al., 2007). Briefly, overnight culture of E. coli containing the pEAL08-2 reporter construct was diluted to OD600nm of 0.05, grown in the presence of quinolones for 2 h, and harvested by centrifugation. P. aeruginosa strains with pEAL08-2 were grown overnight before harvest. Cells were lysed and the LacZ activity was measured using a β-galactosidase enzyme assay kit (Promega, Madison, WI, USA). The LacZ product was measured at 420 nm on a plate reader (BioTek Synergy HT; BioTek, Winooski, VT, USA).
Electrophoretic mobility shift assay (EMSA)
EMSA assays were performed using cell lysates from PAO1, pqs mutants, and Rosetta strains induced for His-SUMO or His-SUMO-PqsR production as described (Cugini et al., 2007). For each sample, 10 µg of cell-lysate was incubated with 0.15 fmol of biotinylated 248-bp pqsA promoter DNA (pqsA’), which was generated by PCR using biotinylated forward primer (TTCTTGCTTGGTTGCCG) and reverse primer (GACAGAACGTTCCCTCTT). Unlabeled pqsA’ probe was generated using the same primers as the biotinylated-pqsA’ without the biotin modification. The reaction mixture contained 10 mM Tris–HCl (pH 8.0), 1 mM EDTA, 50 mM NaCl, 1 mM DTT, 1 µg/µl Poly(dI-dC) and was incubated at room temperature (24 °C) for 20 min. Samples were separated on a 5% polyacrylamide gel in 0.5×Tris Borate EDTA (TBE) run at 100 V for 50 min and transferred onto a positively-charged nylon membrane at 40 V for 1 h. Biotinylated DNA on the nylon membrane was probed by streptavidin-HRP conjugate, detected by a chemiluminescent substrate (Pierce LightShift Chemiluminescent EMSA kit; Thermo Fisher Scientific, Waltham, MA, USA), and visualized by exposing to either an X-ray film or GE ImageQuant RT-ECL.
Saturation transfer difference (STD) NMR
STD NMR experiments were prepared with 1 µM SUMO-PqsR-C87 with increasing concentrations of DHQ (10 nm to 100 µM). Data were collected at 298 K on a Bruker Avance III 600 MHz NMR spectrometer equipped with a 5 mm cryogenically-cooled QC-Inverse probe and using a standard STD pulse sequence with 30 ms 8.4 kHz spin lock to minimize background protein resonances (Kemper et al., 2010). Solvent suppression was achieved using the excitation sculpting scheme. Saturation of the protein signals was performed using a train of 10, 20, or 59 selective 56 dB Gaussian pulses of 50 ms duration with total saturation times of 0.25 s to 5.0 s. The on-resonance frequency was set up at −0.5 ppm. STD spectra were acquired from 64 scans, 2,050 receiver gain, and 14 ppm sweep width.
Quantification of quorum-sensing molecules
Supernatants of overnight cultures of PAO1 grown aerobically or anaerobically were collected and acidified with 0.1% formic acid. RP-HPLC coupled with a mass detector was used to analyze and quantitate QS molecules in culture supernatants. The HPLC-MS system used was a Waters HPLC system equipped with a Waters ZQ Single Quadrapole Mass Detector outfitted with MASS LYNX software. The HPLC/MS method was water/acetonitrile (ACN) gradient with 0.1% formic acid in both solvents. Samples (10 µl) were loaded onto an Ascentis Express C18 column (Sulpeco Analytical, 5 µm particle size, 150mm × 2.1mm) and eluted using an acetonitrile (ACN) gradient at a flow rate of 0.4 ml/min: 30% ACN for 2 min, a linear gradient from 30 to 100% ACN over 30 min, 100% ACN for 5 min. Column temperature was held at 30 °C. The concentrations of QS molecules were determined using calibration curves generated with commercial chemical standards at concentrations ranging from 0.1 µM to 10 mM.
Patient recruitment
This study protocol was approved by the Institutional Review Board of the Medical University of South Carolina, and the study was carried out according to the approved guidelines (IRB number: Pro00012798). Written informed consent was obtained from each patient at the time of recruitment. Patients were selected using the following criteria: over 18 years old, had a positive culture history of P. aeruginosa but negative for Burkholderia species, non-smoker, readily produced sputum, and received routine treatment at the Adult CF Center of the Medical University of South Carolina. Sputum samples were collected during patients’ routine visits at the CF Center clinic and stored at −80 °C until the samples were analyzed.
Quantification of QS molecules in sputum samples
Sputum samples were thawed and digested with 10% sputolysin (Calbiochem, San Diego, CA, USA) in PBS at 37 °C for 15 min. Digested samples were extracted with 3 ml acidified ethyl acetate (0.1% formic acid) twice. The organic layers were pooled and dried under nitrogen. Samples were dissolved in 500 µl methanol and 10 µl sample was analyzed by the HPLC-MS as described above to determine the concentrations of QS molecules in the sputum.
Statistical analysis
The Log-Rank (Mantel-Cox Test) test was used to analyze the C. elegans survival data. A p-value of less than or equal to 0.05 was considered as a significant difference. Other data was analyzed using the Student’s t-test (DF = 1), significant differences were identified with a p value of less than or equal to 0.05.
Results
DHQ maintained P. aeruginosa virulence in C. elegans
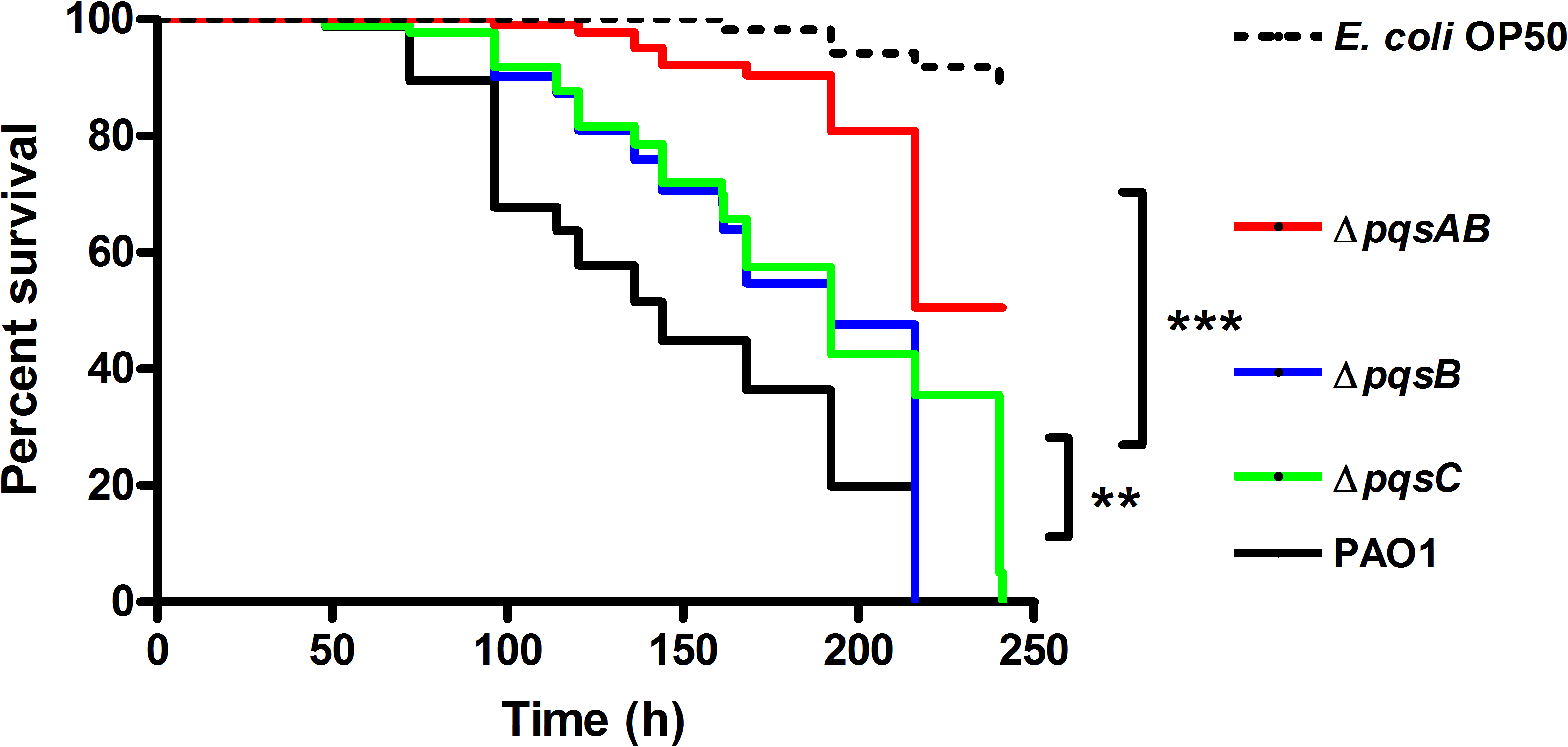
C. elegans is an established infection model to study bacterial pathogenicity in research labs (Korgaonkar et al., 2013; Kwak, Jacoby & Hooper, 2013). P. aeruginosa infections are lethal to the worms due to toxic virulence factors such as pyocyanin and hydrogen cyanide, chelation of iron by PQS, and colonization of the intestinal lumen. To determine the effect of DHQ on P. aeruginosa pathogenicity, we monitored the survival of C. elegans infected with wild-type P. aeruginosa strain PAO1 and pqs mutants (Fig. 1). E. coli OP50 is a food source to maintain the nematodes and served as a non-pathogenic control in our study. PAO1 requires both virulence factor production and colonization of the nematodes to kill C. elegans following several days of incubation (>200 h) (Mahajan-Miklos et al., 1999; Tan, Mahajan-Miklos & Ausubel, 1999; Tan et al., 1999). The Pqs system regulates virulence factor production and plays a role in biofilm formation, both of which play a role in C. elegans killing. Using the DHQ-only producing mutant, ΔpqsB and ΔpqsC strain (Zhang et al., 2008), we were able to examine the effect of DHQ production on virulence compared to a quinolone-null mutant, ΔpqsAB. 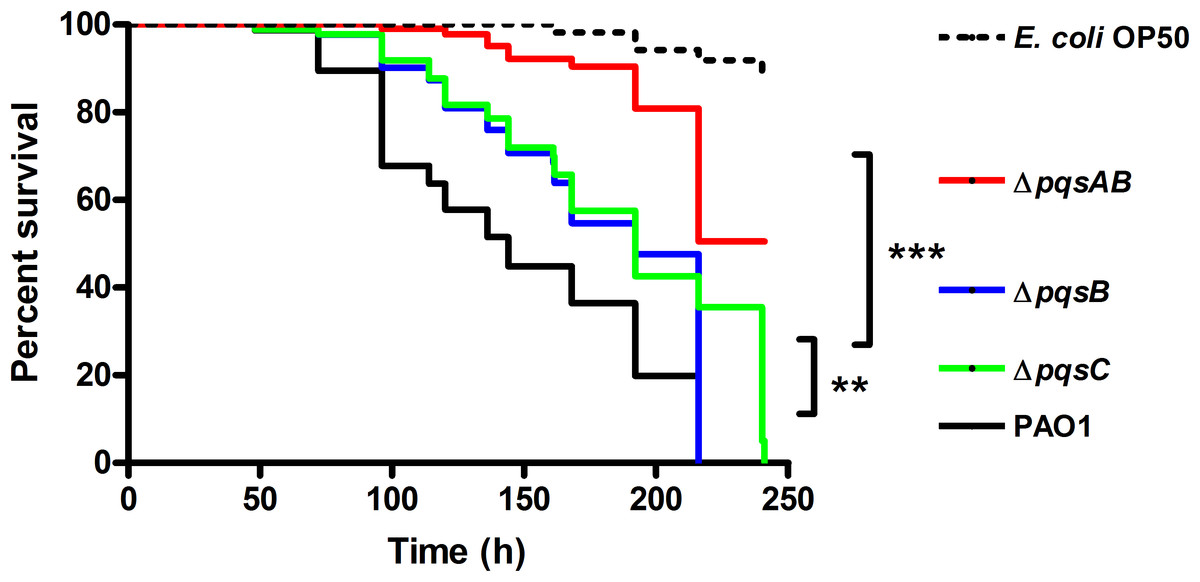
Figure 1: Survival of C. elegans infected with PAO1 and pqs mutants.
P. aeruginosa strains were incubated with C. elegans and worm survival was monitored daily for over 10 days. Each assay contained 20–30 nematodes per strain and was performed in triplicate. Survival data of three independent experiments were combined and plotted in a Kaplan–Meier survival curve. E. coli strain OP50 was used as a non-pathogenic control. Number of worms used for each strains: PAO1 n = 77, pqsAB mutant n = 113, pqsB mutant n = 89, pqsC mutant n = 93, E. coli OP50 n = 115. The survival data were analyzed using the Log-Rank test. Significant p-vulues were indicated by asterisks (∗∗p < 0.001, ∗∗∗p < 0.0001).
{kind=link}
None of the nematodes survived infections from PAO1, ΔpqsB or ΔpqsC, which only produces DHQ, after 220 h of incubation, while 50% of the nematodes infected with ΔpqsAB remained viable (Fig. 1). The quinolone-null mutant ΔpqsAB used in this study was also used in our previous work (Rella et al., 2012), which lacks an internal promoter of the pqs operon. The reduction in virulence of the ΔpqsAB mutant was attributed to the complete loss of quinolone production, while production of DHQ in the ΔpqsB and ΔpqsC mutant maintained pathogenicity towards the nematodes. PqsB and PqsC are both essential for alkylquinolone synthesis, but are not required to produce DHQ (Zhang et al., 2008). C. elegans killing by the ΔpqsB and ΔpqsC mutants showed that DHQ production maintained the same level of virulence in both mutants, which were more virulent against the worms than the quinolone-null mutant ΔpqsAB. Thus, the production of DHQ increased pathogenicity of P. aeruginosa compared to a quinolone-null strain. 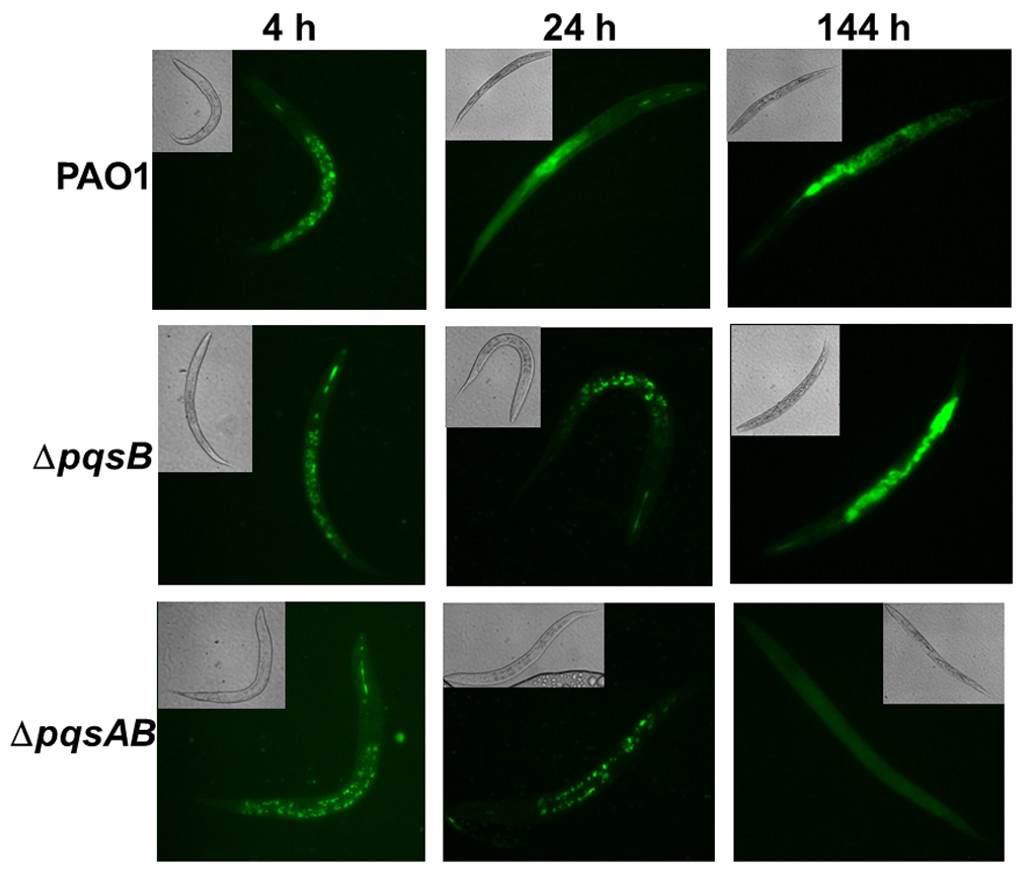
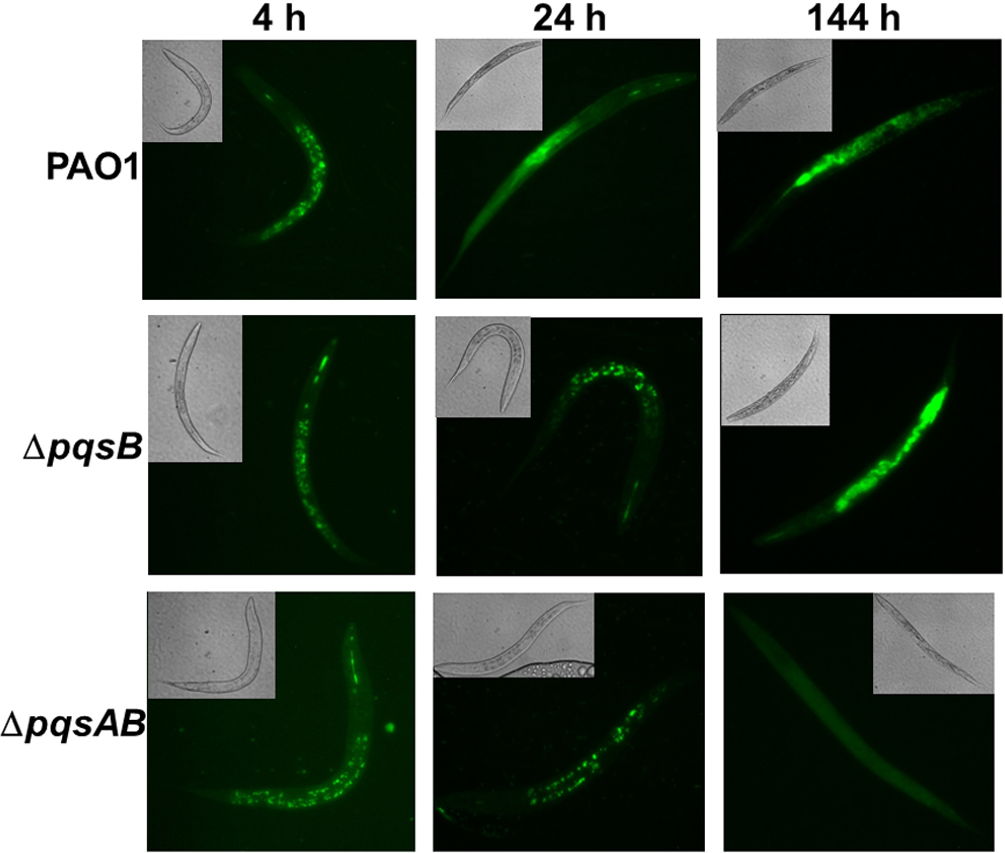
Figure 2: Colonization of C. elegans by P. aeruginosa strains expressing GFP.
Live nematodes were selected to monitor fluorescence from the bacteria. Excitation at 435 nm for 5 s was used for samples from 4 and 24 h, while excitation for 1 s was used at 144 h. Emission was set at 485 nm. 4–6 nematodes per strain were selected from each time point and images from a representative experiment were shown. The GFP-fluorescent signal was quantitated with software ImageJ and the quantified data were shown in Fig. S1.
{kind=link}
Fluorescence-producing bacteria have been used to infect C. elegans to study the internalization of bacteria and their progress through the intestines (Sifri et al., 2003). To visualize colonization of C. elegans by different P. aeruginosa strains, we monitored nematodes infected with PAO1 and the pqs mutants carrying a plasmid that stably expressed GFP (Bloemberg et al., 1997). After 4 h of co-incubation, all nematodes examined contained GFP-expressing bacteria (Fig. 2). After 24 h, GFP-expressing bacteria were distributed throughout the intestinal tract of the nematodes. At 144 h, C. elegans infected with PAO1 and ΔpqsB showed higher GFP fluorescence than those infected with the ΔpqsAB mutant (Fig. 2). We determined the difference in bacterial colonization by comparing fluorescence of the three strains following 1 s of excitation versus 5 s of excitation. A 5 s excitation led to oversaturated GFP signal in nematodes infected with PAO1 and the ΔpqsB mutant, while minimal fluorescence was detected within the ΔpqsAB mutant infected worms. A 1 s excitation resolved the oversaturation in PAO1 and ΔpqsB- infected nematodes and resulted in only background fluorescence from nematodes infected with the ΔpqsAB mutant (Fig. 2). The 144 h was an important time point as it coincided with a sharp decrease in C. elegans survival among the nematodes infected PAO1 and ΔpqsB (Fig. 1). The GFP-fluorescent signals from the infected worms were quantitated using the software ImageJ (Fig. S1). Consistent with the observations on the fluorescent images, no significant difference was detected at 4 h and 24 h post infection. At 144 h time point, worms infected with the pqsAB mutant exhibited significant lower GFP signal than PAO1-infected (p < 0.05) and pqsB-infected worms (p < 0.05). Altogether, our results indicated that production of DHQ increased the ability of P. aeruginosa to colonize and infect C. elegans, which may point to a role in causing infection of a human host.
DHQ supported pyocyanin production in vitro
Virulence factor production is also important for PAO1 infection of C. elegans (Eriksson et al., 2009). Pyocyanin, a blue–green redox-active pigment, is a potent virulence factor that is toxic towards other microorganisms and eukaryotic cells (Lau et al., 2004; Papaioannou, Utari & Quax, 2013; Rella et al., 2012). Both PqsR and PqsE are important for Pyocyanin production in P. aeruginosa: PqsR activates transcription of the pqs operon (Williams & Camara, 2009), PqsE function remains enigmatic until recently being shown to have pathway-specific thioesterase activity and promote alkylquinolone signaling (Drees & Fetzner, 2015).
In order to demonstrate that changes in virulence of the pqs mutants were not due to defects in growth, the growth of the pqs mutants were monitored in LB and synthetic cystic fibrosis medium (SCFM) that was developed based on the nutrient composition of the CF sputum (Palmer, Aye & Whiteley, 2007). The growth curves of the pqs mutants were similar to that of the wild-type PAO1 in both media, demonstrating that these mutants were not defective in replication (Figs. 3A and 3C). Next, we quantified pyocyanin from overnight cultures of PAO1 and pqs mutants grown in LB media. PAO1 produced the highest amount of pyocyanin (about 2.5 µg/ml) followed by the ΔpqsB mutant, which produced 50% of the wild-type level (Fig. 3B). The ΔpqsAB mutants produced the least amount of pyocyanin (Fig. 3B). Nutrient conditions can alter bacterial phenotypes. The SCFM mimics the natural environments of P. aeruginosa and provides less nutrients than LB. Although reduced, a similar trend in pyocyanin production by the strains was observed compared to LB-grown cultures (Fig. 3D). These results support that DHQ production partially maintained pqs signaling and pyocyanin production, whilst alkylquinolones were required for full virulence production. 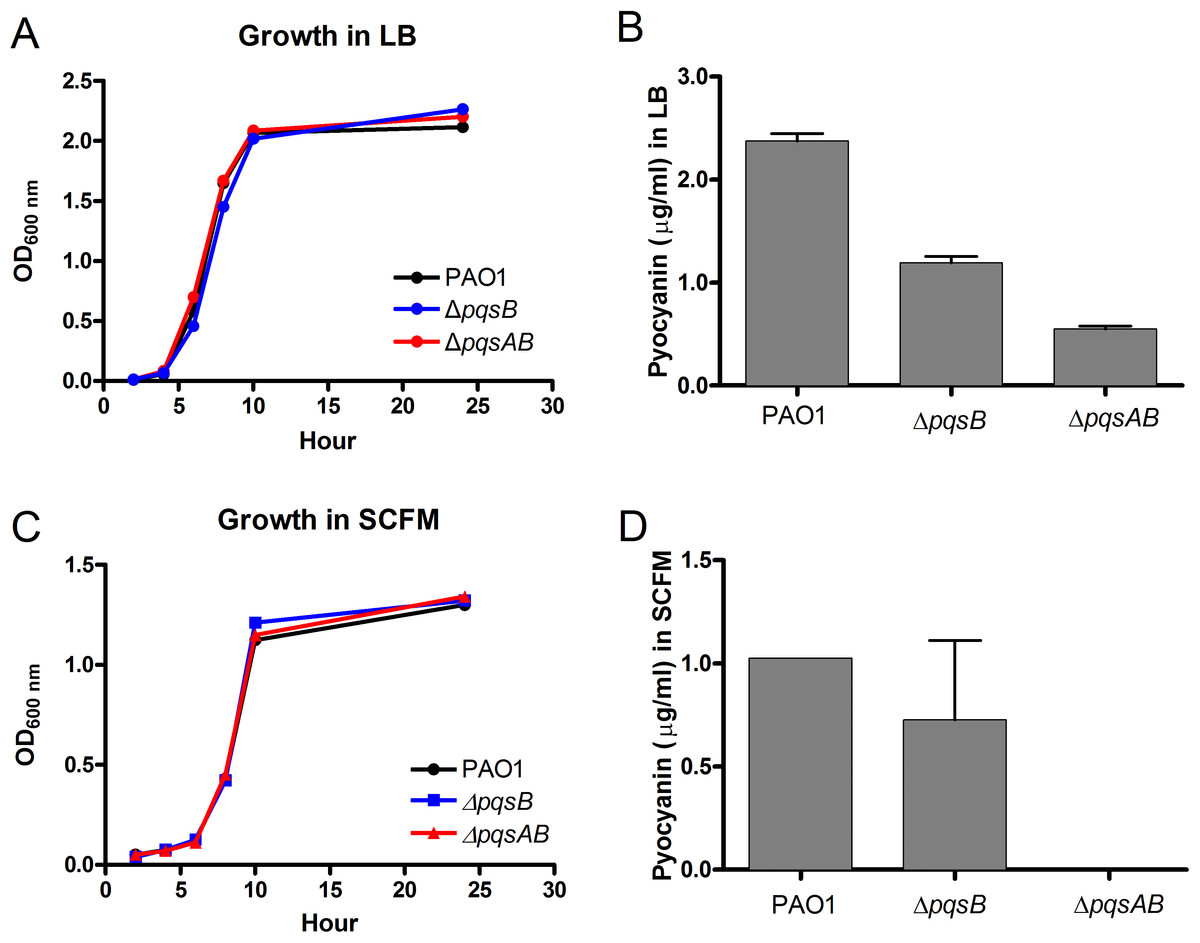
Figure 3: Growth and pyocyanin production by PAO1 and pqs mutants.
(A) Growth curves of P. aeruginosa strains in LB over 24 h. (B) Pyocyanin was extracted from 18-h planktonic cultures grown in LB, quantified spectrophotometrically at 520 nm, presented as mean ±SE from at least three independent measurements. Student’s t-test of pqsB mutant produced significantly higher level of pyocyanin than pqsAB mutant (p-value = 0.0003). (C) Growth curves of P. aeruginosa strains in SCFM over 24 h. (D) Pyocyanin was extracted and quantified from 24 h planktonic cultures grown in SCFM. The pyocyanin from pqsAB mutant in SCFM medium was below the detectable level. Experiments were performed in duplicate and data were presented as mean ± SE.
{kind=link}
DHQ activated PqsR for transcription of the pqs operon
PQS and HHQ activate PqsR for transcription of the pqs operon (Diggle et al., 2007; Dubern & Diggle, 2008). To determine if DHQ affected pqs operon transcription, we used qRT-PCR to monitor the expression of pqsA from PAO1 and pqs mutants using RNA polymerase sigma factor rpoD as the house-keeping gene (Fig. 4). ΔpqsB mutant expressed pqsA at 47% of the wild-type level, while ΔpqsAB mutant showed no detectable pqsA transcript (Fig. 4). Deletion of pqsR also reduced pqsA transcription to basal level, which was consistent with PqsR being an activator of the pqs operon. Deletion of both pqsR and pqsB also resulted in basal level of pqsA transcript (Fig. 4), suggesting that the activation of pqs operon transcription in ΔpqsB was dependent on PqsR. 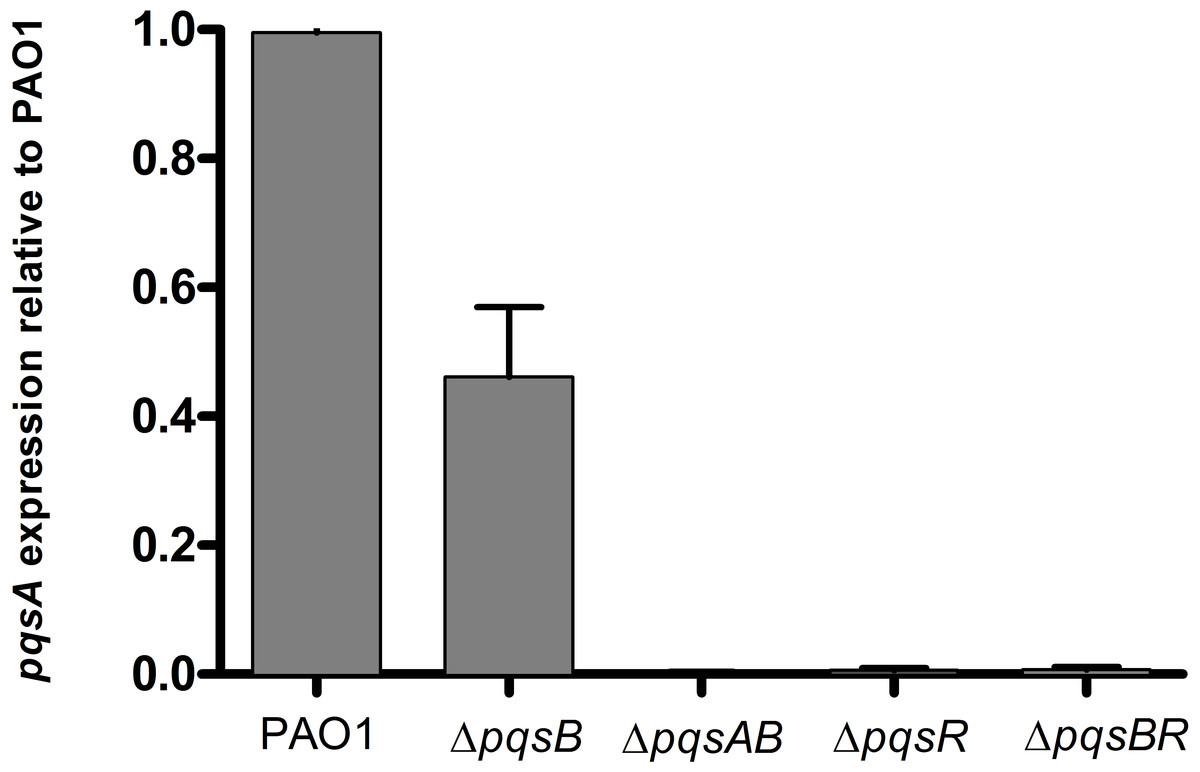
Figure 4: Expression of pqsA in PAO1 and pqs mutants.
Strains grown in LB until cell density reached OD600nm of 1.8. rpoD was used as the house-keeping gene for normalization. Relative abundance of pqsA transcript from pqs mutants compared with the wild-type PAO1 level, which was set as 1. Experiment was performed in triplicate with data shown as mean ± SE. The Student’s t-test analyses showed that pqsA expression level in the pqsB mutant, although lower than the wild-type PAO1 (p-value = 0.008), was statistically significantly higher than pqsAB (p-value <0.0001), pqsR (p-value <0.0001), and pqsBR mutants (p-value <0.0001).
{kind=link}
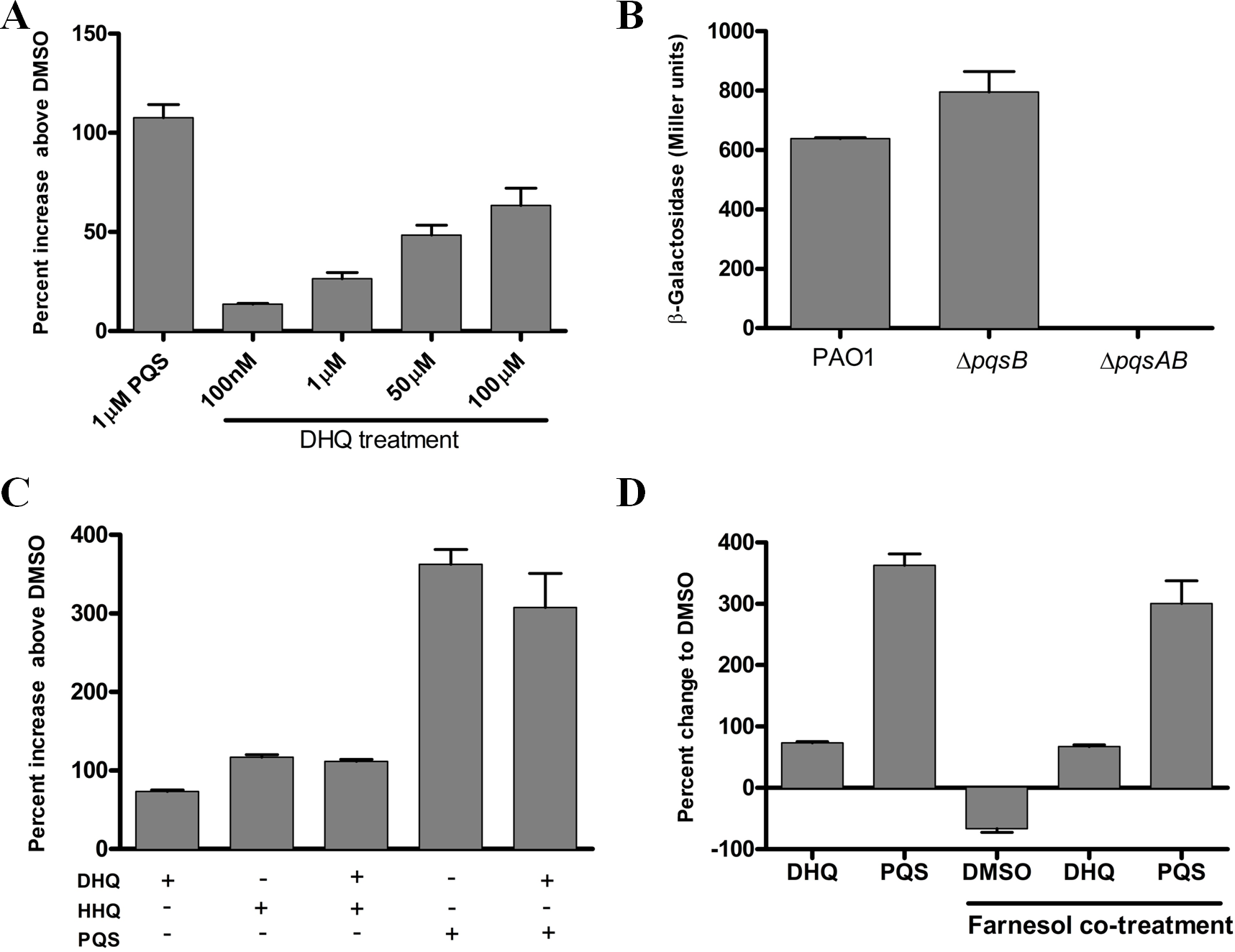
To investigate the effect of exogenous and endogenous DHQ on PqsR activity, we performed promoter-fusion assays in E. coli and P. aeruginosa strains (Fig. 5). The promoter-fusion construct contained the upstream regulatory element (−500 bp) of pqsA fused with the coding sequence of LacZ (Cugini et al., 2007). The reporter construct also contained the pqsR gene, which was controlled by a tac promoter. DHQ supplemented to E. coli containing the reporter construct displayed a concentration-dependent increase in LacZ activity (Fig. 5A). 100 µM DHQ increased the pqsA promoter activity by 60% over the DMSO-treated control, compared with 110% activation by 1 µM PQS (Fig. 5A), suggesting that PQS was more potent in activating PqsR. The effect of endogenous quinolones on PqsR activity was tested in PAO1 and pqs mutants carrying the reporter construct. The pqsA promoter activity from the DHQ-only ΔpqsB mutant was similar to that of PAO1 while the ΔpqsAB strain displayed basal activity (Fig. 5B), indicating endogenous produced DHQ can activate PqsR binding to pqsA promoter. 
Figure 5: Reporter assay of PqsR activity in E. coli and P. aeruginosa.
(A) DHQ supplemented to E. coli carrying the reporter construct pEAL08-2. LacZ activity in cells treated with DMSO was set as the baseline. DHQ increased PqsR activity in a dose-dependent manner whilst PQS was more potent in activating PqsR (p-value = 0.013 from comparing 1 µM PQS and 100 µM DHQ by the Student’s t-test). (B) Overnight cultures of PAO1 and pqs mutants containing the reporter plasmid were used to determine the effect of endogenously generated DHQ on PqsR activation of pqsA promoter. PAO1 and pqsB mutant exhibited similar PqsR activity whereas no detectable activity was observed in the pqsAB mutant, suggesting that endogenously produced DHQ from the pqsB mutant was sufficient to maintain PqsR activity in the absence of alkylquinolones. (C) Co-supplementation of 100 µM DHQ with 30 µM HHQ or 30 µM PQS in E. coli reporter. No statistically significant difference was observed between alkylquinolone (HHQ or PQS) only group and alkylquinolone plus DHQ (HHQ + DHQ or PQS + DHQ) group. (D) Competition of 100 µM DHQ or 30 µM PQS with 250 µM farnesol in the E. coli reporter. DHQ significantly abolished the inhibition of PqsR activity by farnesol (p-value = 0.0023 between DHQ + farnesol treatment and DMSO + farnesol treatment by the Student’s t-test). Data from independent experiments were presented as mean ± SE and analyzed by the nonparametric Student’s t-test.
{kind=link}
To test if DHQ can compete with alkylquinolones for PqsR, DHQ was supplemented in the presence of HHQ or PQS in the E. coli reporter system. DHQ supplemented with either HHQ or PQS did not significantly alter the pqsA promoter activity compared to HHQ and PQS alone (Fig. 5C). Various fungal species produce farnesol as a quorum-sensing molecule, which is also an antagonist of PqsR (Cugini et al., 2007). In the E. coli reporter system, farnesol reduced LacZ activity below the background level (Fig. 5D), consistent with farnesol as an inhibitor of PqsR. Interestingly, co-treatment of farnesol and DHQ alleviated the inhibition of PqsR by farnesol, albeit at a lower level compared with PQS (Fig. 5D). Taken together, endogenous transcription of pqsA from PAO1 and pqs mutants, as well as activity from exogenous DHQ supplemented to the reporter system, show that DHQ can function as a ligand of PqsR to activate the transcription of the pqs operon.
In vitro studies on the interaction of DHQ with PqsR
The interactions between alkylquinolones with PqsR are demonstrated by a recently solved crystal structure of PqsR complexed with its ligand, which shows how ligand binding affects PqsR affinity for its DNA-binding site (Ilangovan et al., 2013). To test how DHQ binding affects PqsR interaction to its DNA binding site, we performed electrophoretic mobility shift assays (EMSA) with biotinylated-pqsA promoter (Bio-pqsA) and cell lysates of PAO1 and the pqs mutants (Fig. 6). Only PAO1 and ΔpqsB cell lysates shifted Bio-pqsA migration, while the ΔpqsAB and ΔpqsR mutants did not show any effect (Fig. 6A). Addition of unlabeled pqsA probe to PAO1 and ΔpqsB cell lysates decreased the amount of shifted Bio-pqsA probe, demonstrating the interaction was specific for pqsA promoter sequence (Fig. 6B). Recombinant PqsR as a fusion protein with N-terminal His-tagged SUMO also showed increased shifted pqsA probe in the presence of PQS or DHQ (Fig. 6C). No pqsA probe was shifted in the control lysate containing His-SUMO tag, demonstrating the shifted pqsA probe was due to PqsR activity specifically (Fig. 6C). The affinity of PqsR for DHQ and PQS was determined semi-quantitatively by EMSA by measuring free Bio-pqsA in response to quinolone supplemented at different concentrations. The calculated Kd of PqsR for DHQ was 150 nM and 33 nM for PQS (Fig. 6D). 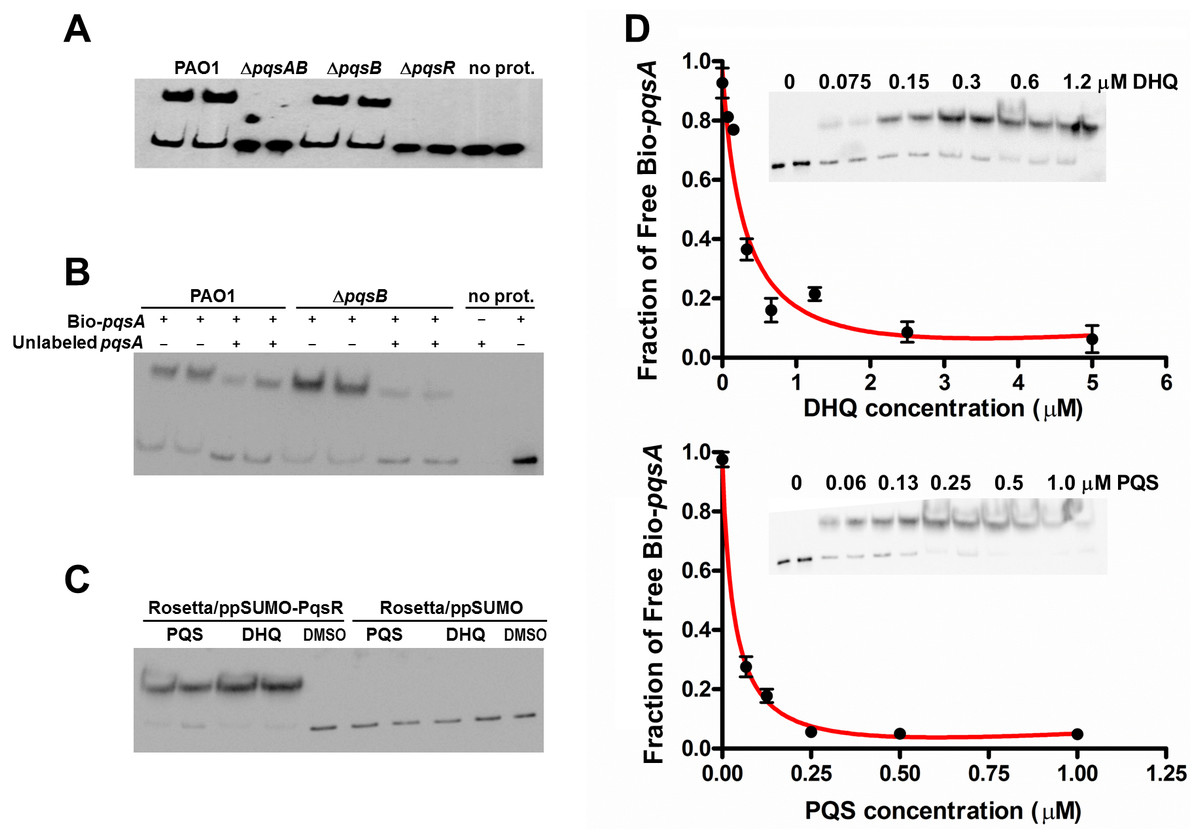
Figure 6: DHQ promoted PqsR binding to pqsA promoter.
(A) EMSA assays with Biotinylated-pqsA (Bio-pqsA) probe incubated with cell-lysates of PAO1 and pqs mutants. (B) The interaction was specific for pqsA promoter as unlabeled-pqsA competed with Bio-pqsA probe. (C) Both DHQ (200 µM) and PQS (30 µM) promoted the interaction between recombinant PqsR with Bio-pqsA. (D) DHQ (0–5 µM) and PQS (0–1 µM) promoted recombinant PqsR binding to Bio-pqsA probe in a concentration dependent manner. Quantification of free Bio-pqsA was performed using ImageJ software and non-linear curve fitting was done using GraphPad Prism software to determine the apparent Kd of PqsR for DHQ and PQS.
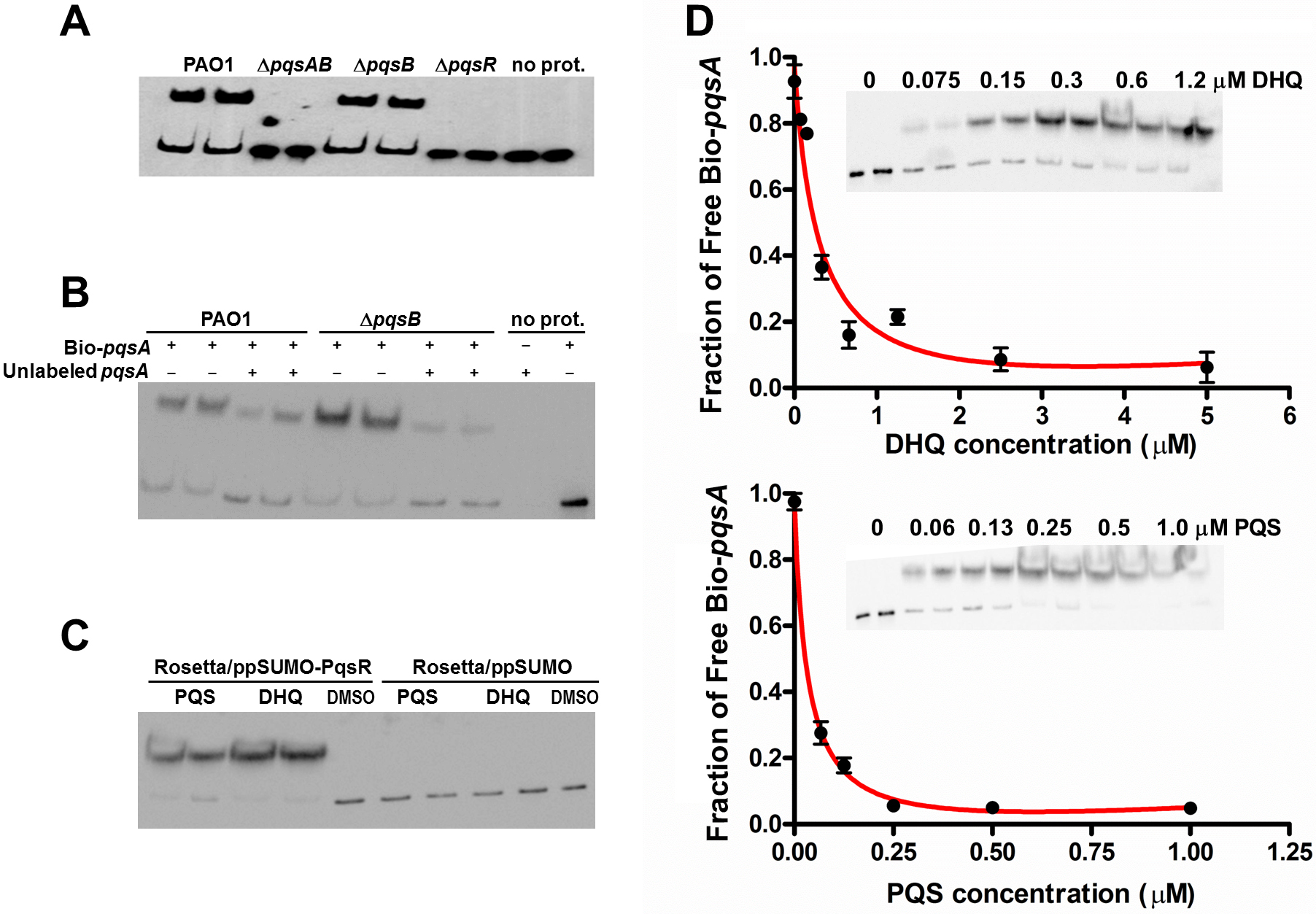{kind=link}
Saturation transfer difference NMR (STD-NMR) detects transient binding of ligand to a protein in solution and was used to study the interaction between DHQ and PqsR. The ligand binding domain of PqsR starting from residue Cys87 (PqsR-C87) was purified as a His-tagged SUMO fusion protein and used in the STD-NMR experiments (Fig. 7). Novel peaks on the PqsR-C87 spectra were quantified for intensity and modeled for single-site binding kinetics (Fig. 7A). The Kd was estimated to be 450 nM (Fig. 7B). Interestingly, the saturation time showed a strong interaction after 3 s, while exhibiting fast-exchange interactions from 0.5 to 2.5 s. The fast-exchange may be explained by hydrophobic interactions within the hydrophobic pocket of PqsR until DHQ was coordinated through hydrogen binding. Using the D-COSY assignments, we mapped the 1-D hydrogen spectra to show that the meta-hydrogen of DHQ participated in hydrogen-binding with PqsR (Fig. 7C). The ortho- and para-hydroxyl groups were not resolved from the water peak. However, we predict those groups were also participating in hydrogen binding based on preliminary docking analyses. 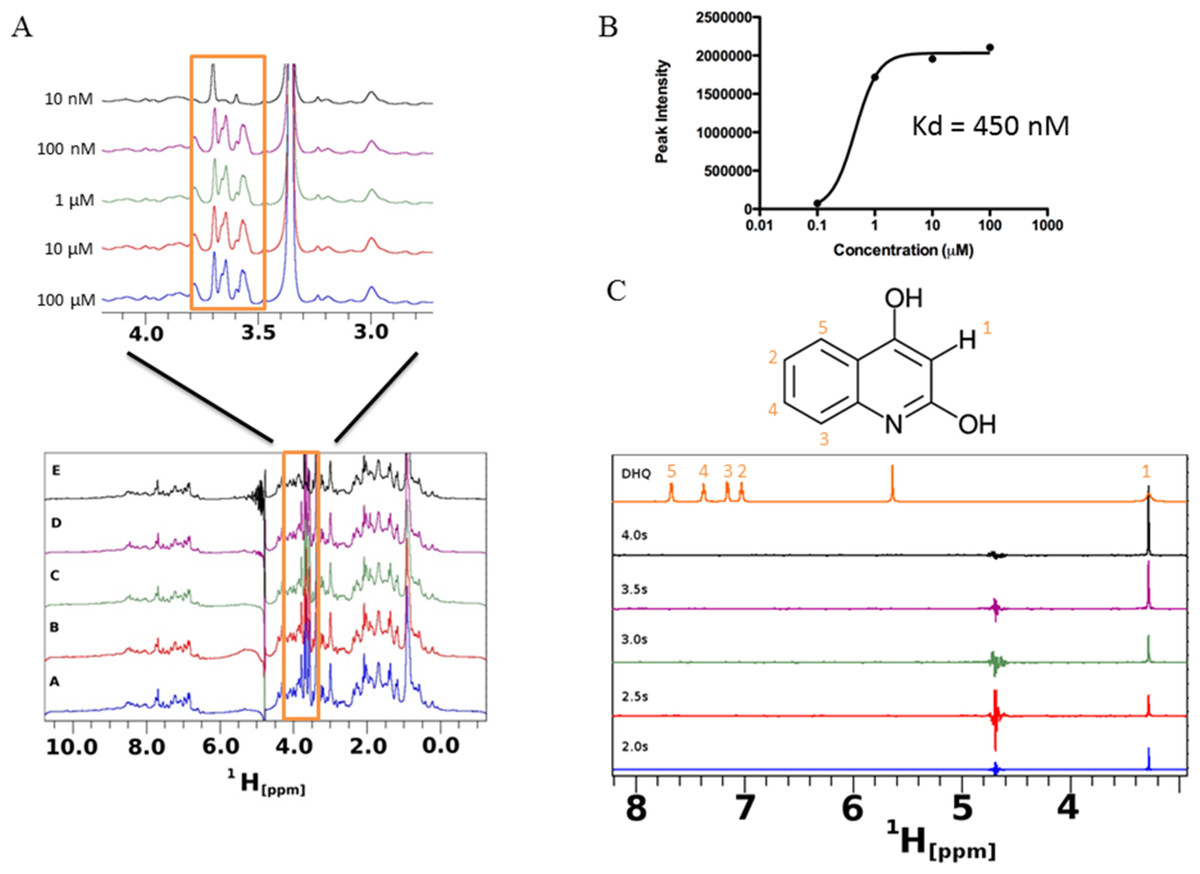
Figure 7: Saturation transfer difference (STD) NMR of SUMO-PqsR-C87 interaction with DHQ.
(A) STD-NMR spectra of 1 µM SUMO-PqsR-C87 titrated with 10 nm to 100 µM DHQ. A close-up view of the changing spectra was used to highlight the differences in peak intensities following increased DHQ concentrations. (B) Peak intensity of the most prominent peak (close-up view) versus concentration of DHQ was used to determine Kd of binding. (C) STD-NMR of 100 µM DHQ added to 1 µM SUMO-PqsR-C87 and measured for saturation times in 0.5 s intervals. Intervals selected to show saturation from initial incubation to 5 s. D-COSY assignments of DHQ used to map interacting hydrogens (numbered in orange).
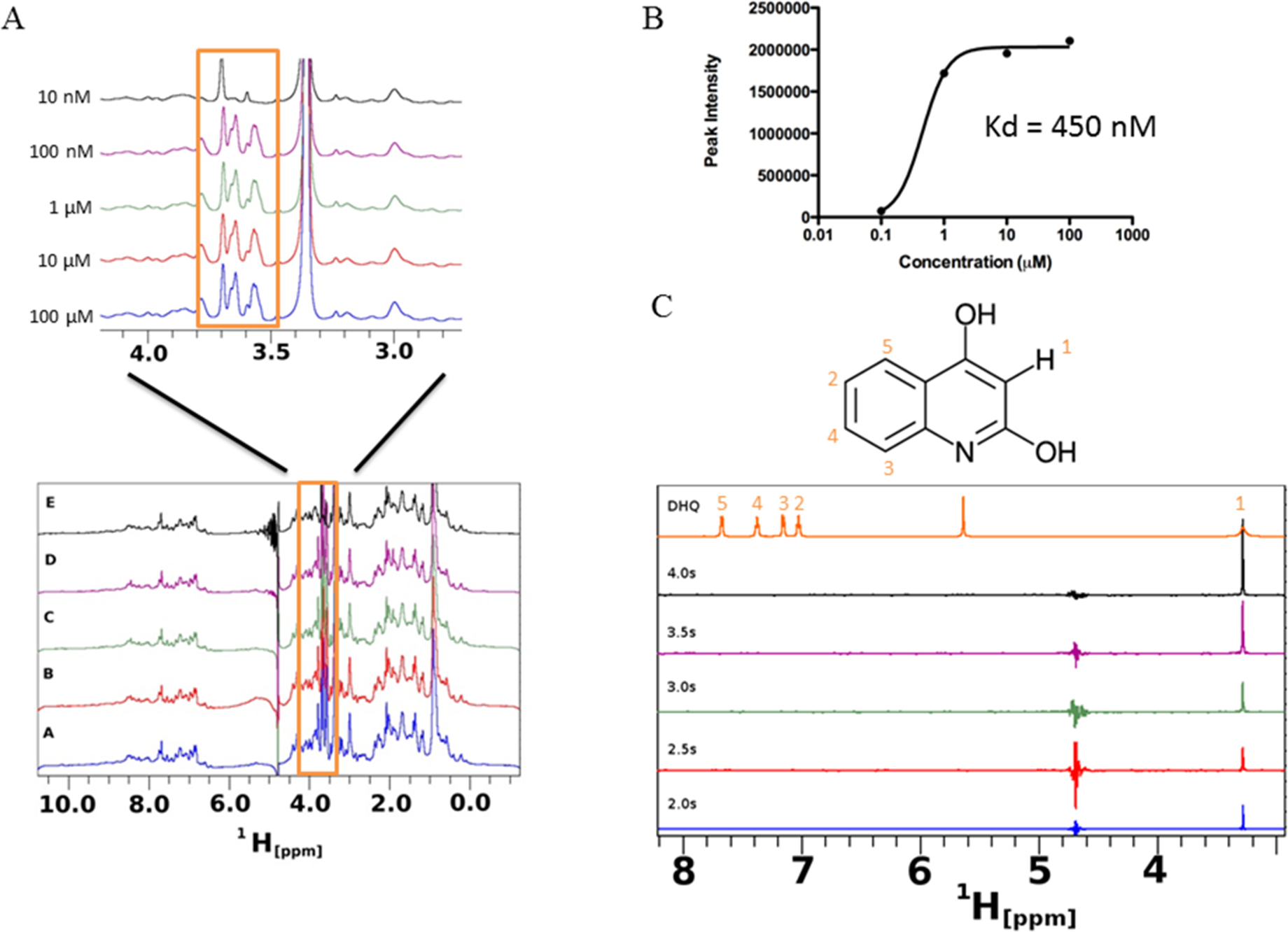{kind=link}
DHQ production under anaerobic conditions
Our results suggested DHQ, similar to PQS and HHQ, promotes P. aeruginosa pathogenesis by activating PqsR; however, conditions found in CF lungs may limit PQS production due to the lack of oxygen (Schobert & Jahn, 2010; Worlitzsch et al., 2002). Further complicating alkylquinolone syntheses is the reduced metabolism in P. aeruginosa biofilms limiting the availability of fatty acid substrates from β-oxidation. As DHQ formation requires smaller, less energy intensive precursors and does not rely on oxygen in the environment, DHQ may play a role apart from the alkylquinolones during chronic colonization of the CF lung. 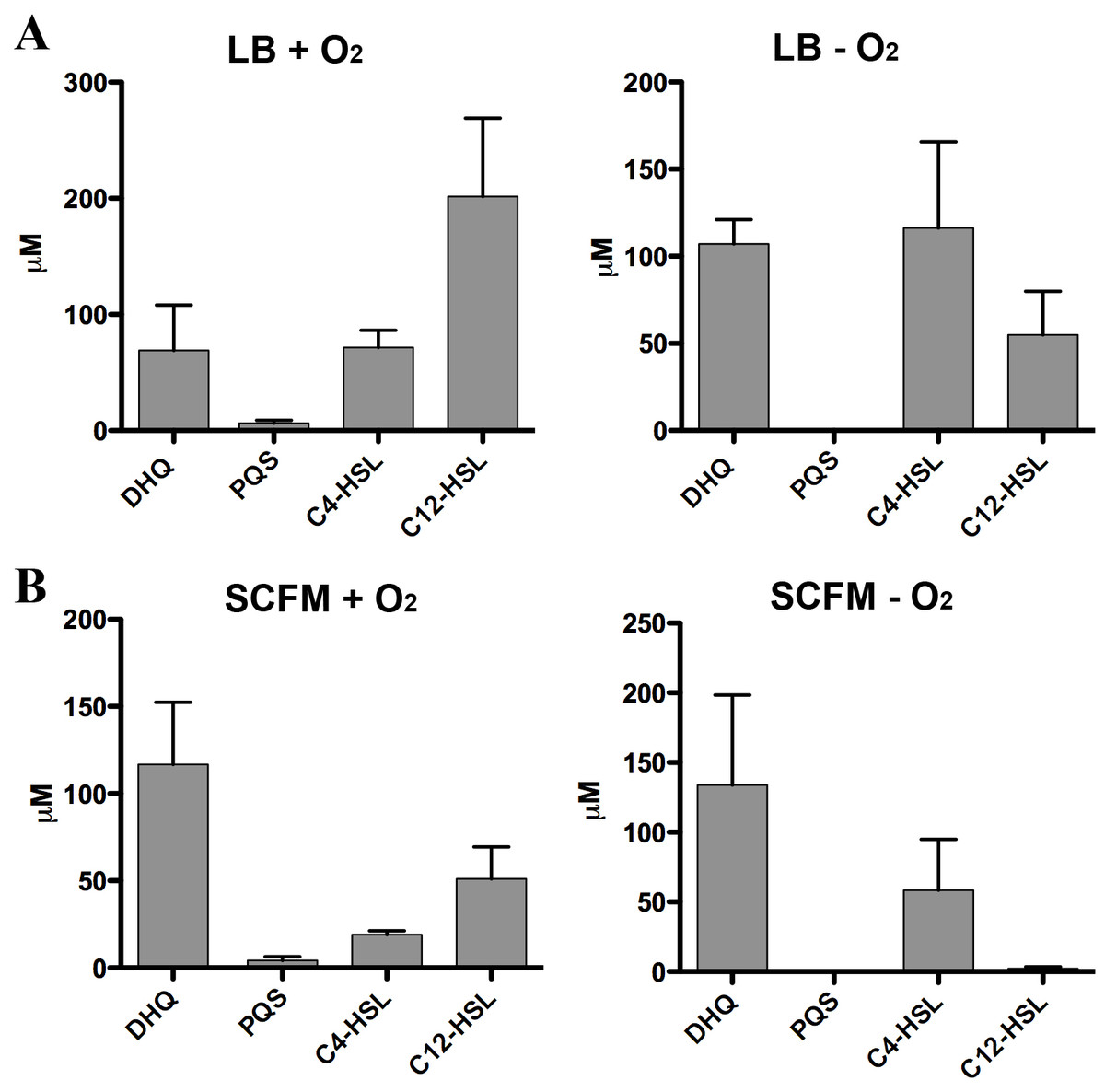
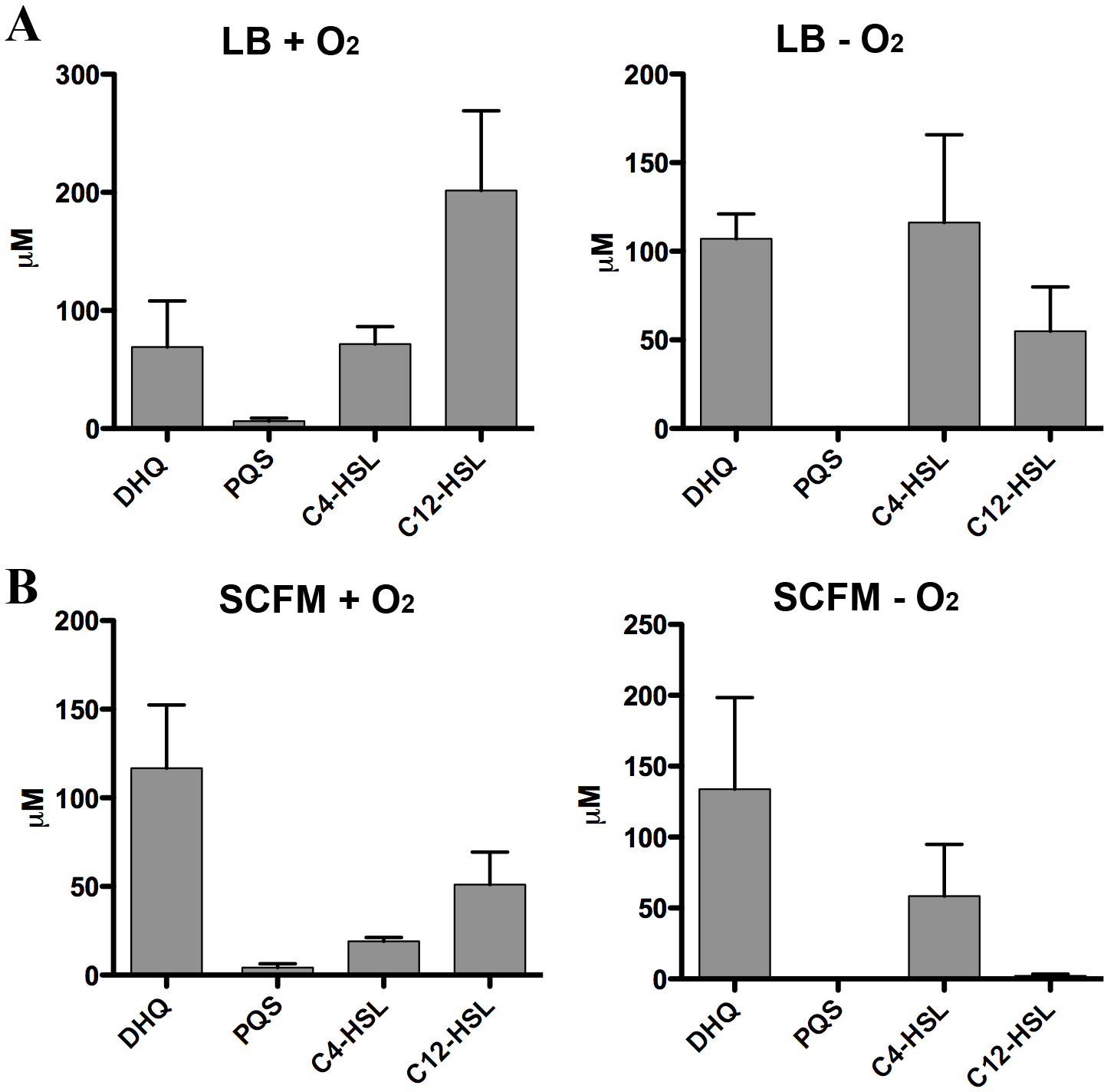
Figure 8: Quantification of extracellular levels of DHQ from aerobic and anaerobic PAO1 cultures.
(A) PAO1 cultures were grown aerobically in LB and anaerobically in LB+400 µM sodium nitrate until the culture reached OD600 of 2.0. Culture supernatants were acidified with formic acid and QS molecules were analyzed using HPLC-MS. The concentrations were determined using calibration curves of QS standards. (B) PAO1 cultures in SCFM were treated in the same manner and grown aerobically and anaerobically to determine the concentrations of QS molecules. Data represented as mean ± SE from at least two independent experiments.
{kind=link}
Strict anaerobic cultures do not produce PQS and express lower pqs operon activity compared to aerobic cultures (Lee et al., 2011). To determine how DHQ concentration is affected during anaerobic growth, we quantified QS molecules from PAO1 cultures grown aerobically and anaerobically (Fig. 8). Concentration of C4-HSL was higher than 3-oxo-C12-HSL in the anaerobic samples, consistent with the relative abundance of the two HSL signals under anaerobic conditions as found in the CF lungs (Hassett et al., 2009). High levels of extracellular DHQ were detected from cultures grown in LB both in the presence and absence of oxygen (Fig. 8A). Interestingly, anaerobic culture supernatants contained higher concentrations of DHQ compared to aerobic cultures in LB. As expected, PQS was not detected under anaerobic conditions. Similar trends of QS molecules were observed from PAO1 cultured in SCFM aerobically and anaerobically (Fig. 8B). Comparison of the DHQ concentrations between LB and SCFM showed that DHQ level was higher in the SCFM, but the difference was not statistically significant (Fig. 8). Together, these results showed DHQ was the predominant quinolone produced in both rich and SCFM medium regardless of the oxygen level. The production of DHQ in anaerobic condition suggest a specific function of DHQ under low oxygen environments where PQS production is limited; however, further research is needed to understand P. aeruginosa transcriptional regulation in hypoxic conditions and the dynamics of QS molecules within the anaerobic mucous.
Detection of DHQ in CF patient sputa
Alkylquinolones have been detected in the CF patient samples (Collier et al., 2002). We analyzed the sputum samples from stable CF patients to determine whether DHQ was detectable in the CF lungs. Sputum samples were collected during CF patient routine clinic visits and were analyzed by HPLC-MS for DHQ concentrations. More than 80 sputum samples from 45 patients were collected and analyzed. DHQ was readily detected in 34 sputum samples, and the concentrations covered a wide range. The median of DHQ concentration in stable CF sputa was 123 µM with an interquartile range between 25 and 464 µM. The wide range of DHQ concentrations in the patient samples reflected the non-uniform nature of the sputum samples. Nonetheless, the presence of DHQ in the patient samples demonstrated that DHQ was produced during an infection in the CF lungs, and the effects of long-term exposure to high concentration of DHQ on host cells remains to be determined.
Discussion
Alkylquinolones are well characterized for their role in activating the Pqs signaling system as a ligand of the transcription factor PqsR. Our work provides the first evidence that DHQ also binds to PqsR to activate the transcription of the pqs operon. Transcriptional studies and EMSA experiments demonstrated that DHQ, though not as potent as PQS, activated PqsR for increased transcription of the pqs operon. STD-NMR determined the interaction between DHQ and PqsR as well as the binding affinity in vitro. HPLC-MS analyses of P. aeruginosa culture supernatants showed that DHQ was present in high extracellular concentrations in both rich medium and SCFM regardless of oxygen availability. Within chronically infected patient samples, DHQ was detected in the CF patient sputum, suggesting DHQ as an extracellular effector to aid P. aeruginosa in the disease environment. Finally, we have established that production of DHQ maintained pathogenicity using the C. elegans infection model. Specifically, production of DHQ resulted in increased colonization and pyocyanin production compared with the quinolone-null mutant.
Among the three well-known QS systems in P. aeruginosa, the Pqs system was most recently identified (Pesci et al., 1999). Some aspects of the Pqs system remain to be elucidated, including the activities of specific enzymes in the biosynthesis of alkylquinolones (Drees & Fetzner, 2015; Dulcey et al., 2013) and the transcriptional regulation of the pqs operon (Brouwer et al., 2014; Dotsch et al., 2012; Knoten et al., 2014). Four transcriptional start sites (TSSs) have been identified in the pqs operon, two in the promoter region of pqsA (pqsA−339 and pqsA−71), the third upstream of pqsB (pqsB−31) (Dotsch et al., 2012), and the fourth just upstream of the pqsC stop codon (Knoten et al., 2014). The Bio-pqsA probe used in this study corresponded to the upstream segment of pqsA−71 TSS. Both DHQ and PQS can bind to PqsR to activate the pqs operon transcription from the pqsA−71 TSS. RhlR was recently shown to regulate the longer transcript of the pqs operon from the pqsA−339 TSS by forming a hairpin structure in the 5′-untranslated sequence (Brouwer et al., 2014). Nothing is known about the regulation of the two internal TSSs. Future work is needed to identify transcriptional regulators of the alternate transcripts of the pqs operon. A putative regulatory site was identified in the second internal transcript (Knoten et al., 2014) and it will be interesting to determine if DHQ or alkylquinolones function as ligands for such regulators.
Apart from its role in transcriptional regulation, the importance of DHQ under anaerobic conditions should garner interest because low-oxygen condition is encountered by colonizing P. aeruginosa during a chronic infection. Our results showed that DHQ was secreted in high concentration regardless of oxygen availability. For anaerobic growth conditions, nitrate acts as an alternate electron acceptor in P. aeruginosa (Hassett, 1996); however, PQS inhibits nitrate-respiratory chain activity through iron chelation (Toyofuku et al., 2008). PQS is secreted via membrane vesicles, which are formed under both aerobic and denitrifying condition (Toyofuku et al., 2013). HHQ is secreted by the RND-type efflux pump MexEF-OprN, which is up-regulated under anaerobic conditions (Fetar et al., 2011; Lamarche & Deziel, 2011). Our preliminary work has also indicated that DHQ was secreted from the same efflux pump. This mechanism of secretion may therefore explain why DHQ was present in high levels in the oxygen-limiting CF sputum. As CF patients are chronically infected by P. aeruginosa, more work is required to determine the effects of long-term exposure of lung cells to DHQ.
Taken together, our findings support that DHQ plays similar multifactorial roles compared to the alkylquinolones in P. aeruginosa pathogenicity. DHQ is present at high concentrations in both aerobic and anaerobic conditions, which is significant given the nature of low-oxygen in the lungs of CF patients. However, we have only begun to understand how the quinolones function individually. Future efforts should focus on how DHQ interacts with other quinolones and extracellular molecules and how these interactions affect P. aeruginosa pathogenicity. It will also be interesting to determine how DHQ affects the fitness of P. aeruginosa in its natural environments such as soil and water and how DHQ affects other microorganisms cohabitating with P. aeruginosa.
Supplemental Information
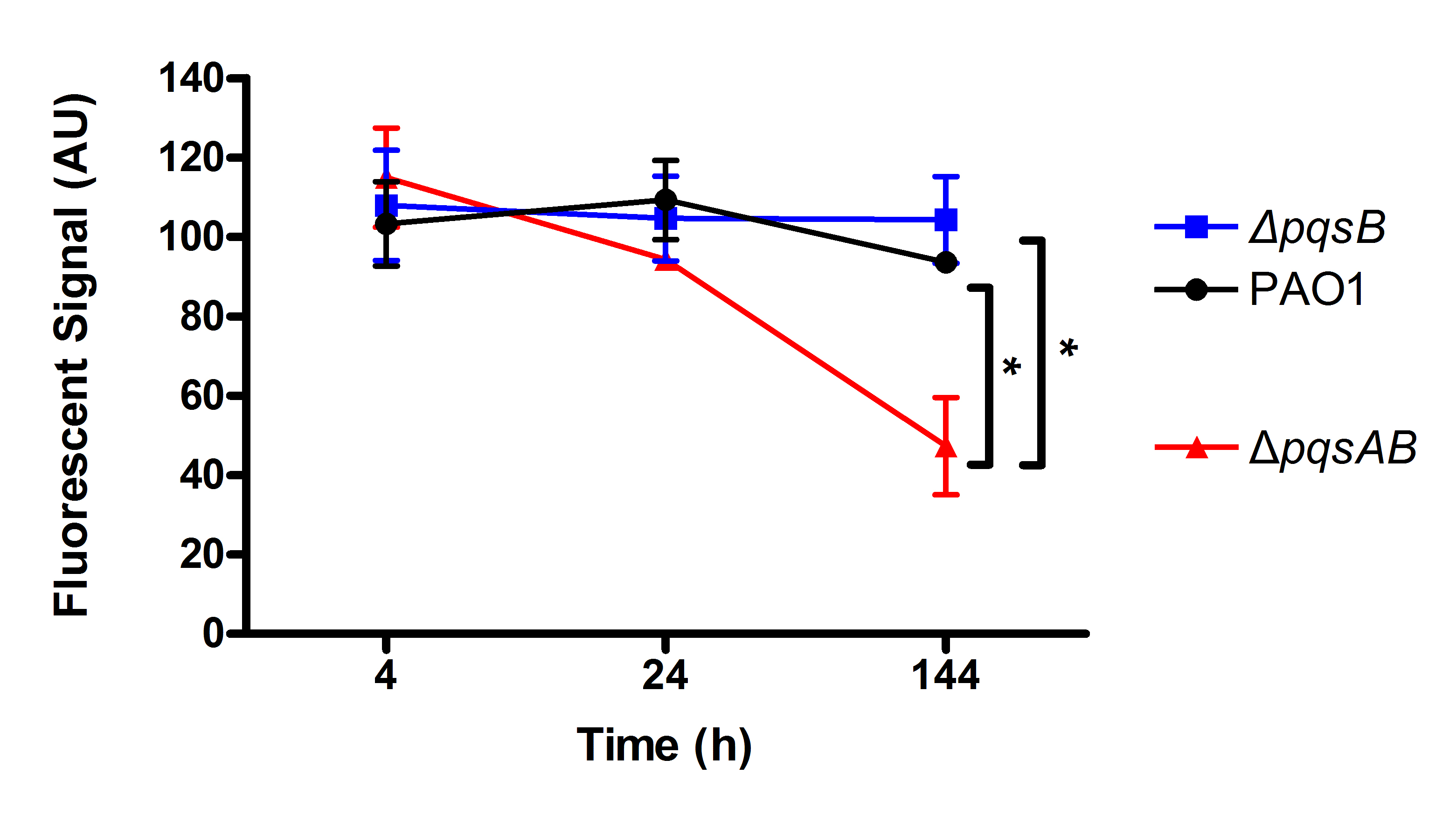
Quantified GFP-fluorescence from colonized worms.
Similar amount of GFP signal was detected in the worms infected with different strains at 4 h and 24 h post infection. At 144 h time point, pqsAB mutant showed significantly reduced GFP signal (∗p-value <0.01), indicating decreased colonization of the worms by pqsAB than PAO1 and pqsB mutant.
{kind=link}
Original images of PAO1 in Fig. 2
Original images from phase contrast and fluorescent microscopy of wild-type PAO1, which were used to generate Fig. 2.
Original images of pqsB mutant in Fig. 2
Original images from phase contrast and fluorescent microscopy of pqsB mutant, which were used to generate Fig. 2.
Original images of pqsAB mutant in Fig. 2
Original images from phase contrast and fluorescent microscopy of pqsAB mutant, which were used to generate Fig. 2.
Raw data for figures
Each figure’s raw data are shown in a spreadsheet, which is labeled to show the corresponding figure.