
Selective constraint acting on TLR2 and TLR4 genes of Japanese Rana frogs
- Published
- Accepted
- Received
- Academic Editor
- Joseph Gillespie
- Subject Areas
- Evolutionary Studies, Molecular Biology
- Keywords
- Amphibian, Ranidae, Toll-like receptors
- Copyright
- © 2018 Lau et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2018. Selective constraint acting on TLR2 and TLR4 genes of Japanese Rana frogs. PeerJ 6:e4842 https://doi.org/10.7717/peerj.4842
Abstract
Toll-like receptors (TLRs) are an important component of innate immunity, the first line of pathogen defence. One of the major roles of TLRs includes recognition of pathogen-associated molecular patterns. Amphibians are currently facing population declines and even extinction due to chytridiomycosis caused by the Batrachochytrium dendrobatidis (Bd) fungus. Evidence from other vertebrates shows that TLR2 and TLR4 are involved in innate immunity against various fungi. Such genes therefore may play a functional role in amphibian-chytridiomycosis dynamics. Frogs from East Asia appear to be tolerant to Bd, so we examined the genetic diversity that underlies TLR2 and TLR4 from three Japanese Ranidae frog species, Rana japonica, R. ornativentris and R. tagoi tagoi (n = 5 per species). We isolated 27 TLR2 and 20 TLR4 alleles and found that these genes are evolutionarily conserved, with overall evidence supporting purifying selection. In contrast, site-by-site analysis of selection identified several specific codon sites under positive selection, some of which were located in the variable leucine rich repeat domains. In addition, preliminary expression levels of TLR2 and TLR4 from transcriptome data showed overall low expression. Although it remains unclear whether infectious pathogens are a selective force acting on TLRs of Japanese frogs, our results support that certain sites in TLRs of these species may have experienced pathogen-mediated selection.
Introduction
Toll-like receptors (TLRs) are a type of pattern recognition receptor that recognize pathogen-associated molecular patterns (PAMPs) such as bacterial cell walls and nucleic acids (Medzhitov, 2001). The signalling of TLRs triggers the synthesis and release of pro-inflammatory cytokines, and thus TLRs have an important role in innate immunity and activation of adaptive immunity. TLRs are type 1 membrane glycoproteins comprised of extracellular and cytoplasmic domains; the extracellular domain is also considered as a ‘pathogen-recognition domain’ with a variable number of leucine rich repeats (LRR, protein motifs in the ectodomain inferred to be important for recognising molecules), while the cytoplasmic signalling domain is a conserved toll/IL-1 domain (Mikami et al., 2012). The TLR repertoire differs between vertebrate groups ranging from 10 loci in humans and great apes to 21 in urodele amphibians (Takeda, Kaisho & Akira, 2003; Quach et al., 2013; Babik et al., 2014). Specific TLR loci are generally considered to recognize different groups of PAMPs, for example, TLR1 and TLR6 associate with TLR2 to recognize acylated lipopeptides, TLR4 recognize lipopolysaccharides in gram-negative bacteria, and TLR5 recognize bacterial flagellin (Poltorak et al., 1998; Voogdt et al., 2016). In addition, TLR2 and TLR4 can recognize PAMPs derived from different fungi species (reviewed by Roeder et al., 2004; Luther & Ebel, 2006).
In amphibians, TLRs have been described in Xenopus frogs (20 loci from 14 families, Ishii et al., 2007) and newts (21 loci from 13 families, Babik et al., 2014). TLR families characterized in both frogs and newts include TLR01, TLR02, TLR03, TLR05, TLR07, TLR08, TLR09, TLR12, TLR13, TLR14, TLR21, and TLR22. However, the prevalence of TLR4 across amphibian taxa is uncertain: no TLR4 orthologs were found in newts, but putative TLR4 were identified in Xenopus (Ishii et al., 2007; Babik et al., 2014). In addition, TLR4 was one of the 11 TLR genes that were isolated from transcriptome data of Bombina maxima frogs (Zhao et al., 2014).
Most TLR genes are functionally constrained, and typically have high sequence conservation and slow evolutionary rates to maintain a functional role of recognizing conserved PAMPs (Roach et al., 2005), although extracellular LRR domains have higher evolutionary rates compared to intracellular domains (Mikami et al., 2012). An overall signature of purifying selection was identified in TLRs of urodele amphibians, although a few individual codons were found to be evolving under positive selection (Babik et al., 2014), as has been observed in other vertebrates (Wlasiuk & Nachman, 2010; Shang et al., 2018). The identification of TLR-disease associations in vertebrates (Tschirren et al., 2013; Noreen & Arshad, 2015) also supports that episodic selection can occur in TLR genes as a response to changes in pathogen diversity. In addition, human TLR4 displayed significantly negative Tajima’s D values in nonsynonymous variants (Smirnova et al., 2001), and subsequent evidence supporting selection for rare TLR4 variants was found (Smirnova et al., 2003). Therefore, while TLRs may be under functional constraints, there is evidence across vertebrates that positive selection may act on these genes in response to local pathogens.
Chytridiomycosis is a disease in amphibians caused by the fungal pathogen Batrachochytridium dendrobatidis (Bd). This disease has been linked to the decline of amphibian populations worldwide (Daszak, Cunningham & Hyatt, 2003; Longcore et al., 2007; Skerratt et al., 2007; Wake & Vredenburg, 2008). Despite Bd being prevalent in Korea and Japan (Goka et al., 2009; Bataille et al., 2013), within endemic East Asian frogs there is no evidence of Bd-related declines and no published reports of Bd susceptibility following experimental infection; this supports that such frogs could be Bd-tolerant. Additionally, genetic evidence for high Bd genetic diversity and endemism in this region indicates that Bd is endemic to Asia (Fisher, 2009; Bataille et al., 2013), suggesting a long co-evolutionary history between the Bd pathogen and Asian amphibians.
While adaptive immune genes of Japanese frogs have been studied in the context of diseases like chytridiomycosis (Lau et al., 2016; Lau et al., 2017), there are limited studies involving innate immunity genes including TLRs. Since TLR2 and TLR4 have been shown to play a role in innate immune responses to various fungi (Roeder et al., 2004; Luther & Ebel, 2006), TLRs therefore may be involved in Bd resistance (Richmond et al., 2009).
Basal expression of TLRs can provide an overview of the function of these genes, and have been examined in a few anuran species including Bombina and Xenopus frogs. In B. maxima, expression levels of TLR2 and TLR4, measured using quantitative RT-PCR, were variable between different adult tissues (Zhao et al., 2014). In X. laevis, ubiquitous expression of both TLR2 and TLR4 was detected in adults and tadpoles using standard PCR, but expression levels were not quantified (Ishii et al., 2007). This current study provides the opportunity to expand the knowledge about basal TLR expression in amphibians.
To better understand the gene complements of anuran innate immunity, here we characterize the genetic diversity and selection patterns of two candidate TLR genes in three Japanese Rana species using molecular cloning and sequence analyses. Given the potential immunological importance of TLRs against fungal pathogens, we hypothesized that TLRs would be subjected to purifying selection in species which show marked resistance to Bd. In addition, we also conducted preliminary investigation of TLR expression using published transcriptomic data, to determine whether basal expression is similar across different life stages and tissue types.
Methods
Animals
All sample collection was approved by Hiroshima University Animal Research Committee, approval number G14-2. Adult skin tissues were collected from three common Ranidae frog species from Japan (n = 5 per species): the Japanese brown frog (Rana japonica), the montane brown frog (Rana ornativentris), and Tago’s brown frog (Rana tagoi tagoi). All frogs are the same individuals used previously to characterize MHC class I and II genes (Lau et al., 2016; Lau et al., 2017, Table S1). All animals were housed in laboratory conditions for a minimum of five weeks and exhibited no clinical signs of disease prior to euthanasia, and thus considered ‘healthy’. Animals were euthanized through immersion in tricaine methanesulfonate (MS222, 0.5–3 g/L water), and preserved in RNAlater (Applied Biosystems, Carlsbad, CA, USA) at −20 °C prior to excision of skin sample and genomic DNA (gDNA) extraction using DNAsuisui-F (Rizo Inc., Tsukuba, Japan) following manufacturer’s protocol.
Isolation of TLR genes from transcriptome data set and primer design
To isolate TLR2 and TLR4 genes, we utilised the published transcriptomic data set previously compiled using Illumina sequences from immune tissues of the three species (Lau et al., 2017). Briefly, we used the assembled transcripts that were annotated with NCBI-BLAST-2.3.30 against the Swissprot protein database (https://www.expasy.ch/sprot), and isolated all transcripts that had top hits from BLAST search to published TLR genes of other vertebrates. We manually scanned the NCBI-BLAST search results and aligned each transcript with orthologous genes from Xenopus laevis and Nanorana parkeri (Xenbase, http://www.xenbase.org/, RRID:SCR_003280, and GenBank accession numbers XM_002933491, XM_018557931, XM_018232906, XM_018565865). Due to low coverage of sequence data, full-length contigs were not available for TLR2 and TLR4 genes in all three species (Figs. S1 and S2); in such cases, fragmented contigs were used in the alignment. The genomic structure of most vertebrate TLR genes are unique in that the majority of their coding sequence is located within a single exon. From the alignments, we used Primer 3 (Rozen & Skaletsky, 1998) to design degenerate primers that amplified 2348 bp and 2072 bp fragments within a single exon of TLR2 (RanaTLR2_F: 5′-TGRTTGCATACATATGGAGTTG-3′, RanaTLR2_R: 5′-GTGGTCCTCTGGCTGAAGAG-3′) and TLR4 (RanaTLR4_F: 5′-CTGGCAAGCCTTT CTGAACT-3′, RanaTLR4_R: 5′-AGCGGARCATCAACTTTACG-3′), respectively, across all three species (Table S1).
TLR PCR and sequencing
Polymerase chain reaction (PCR) amplification was conducted in Applied Biosystems® Veriti® thermal cycler in 10 µL reactions with 0.25 U TaKaRa Ex Taq® polymerase (Takara Bio Inc., Kusatsu, Shiga, Japan), 1x Ex Taq PCR buffer, 0.2 mM each dNTP, and 0.7 µM each primer and 0.5–1.0 µL skin gDNA samples (n = 5 per species) with the following cycle condition: initial Taq activation at 95 °C for 1 min, then 35 cycles of 30-s denaturation at 95 °C, 30-s annealing at 60 °C (TLR2) or 61 °C (TLR4), and 80-s extension at 72 °C, then a final extension of 72 °C for 3 min. Since TLR alleles could not be phased in heterozygous individuals by sequencing alone, we used molecular cloning followed by Sanger sequencing. PCR products were ligated into T-Vector pMD20 (Takara Bio Inc., Kusatsu, Shiga, Japan) using DNA Ligation Kit 2.1 (Takara Bio Inc.) and incubated for 30 min at 16 °C. For cloning, ligation reactions were transformed into JM109 competent cells (Takara Bio Inc., Kusatsu, Shiga, Japan) and cultured on selective LB plates containing 50 µg /mL ampicillin overnight at 37 °C. We then amplified positive clones (4 –10 per individual reaction) using M13 primers and similar PCR conditions, and purified using ExoSAP-IT® (Affymetric Inc., Santa Clara, CA, USA). As amplicons were over 2 kbp length, we utilised four to six additional sequencing primers (Table S2, Figs. S3 and S4) in addition to M13 primers for sequencing with BigDye® Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) and ABI 3130xl automated sequencer.
Sequence analyses, dN∕dS comparison with other genes, and selection tests
We measured polymorphism and divergence of the TLR2 and TLR4 sequences using DnaSP 6.10.03 (Rozas et al., 2017), including number of segregating sites (S), number of alleles (NA), average number of nucleotide differences (k), nucleotide diversity (π), Tajima’s D (D) and normalized Fay and Wu’s H (Hn). We calculated synonymous (dS) and nonsynonymous (dN) divergence and the ratio (dN∕dS) between the focal species using MEGA7 (Kumar, Stecher & Tamura, 2016). We then compared the dN∕dS ratio with that of major histocompatibility complex (MHC) class I and II (average among the three species), which are known to be under balancing selection in these species (Lau et al., 2016; Lau et al., 2017). In addition, we compared with dN∕dS ratio of all orthologous genes collated from the transcriptome data set of Lau et al. (2017). This consisted of over 3,000 orthologous amino acid sequences from each of the three species that were identified using Proteinortho V5.15 (Lechner et al., 2011). We then extracted nucleotide coding sequences of orthologous genes from transcriptome data sets using a custom python script, and used PhyloTreePruner (Kocot et al., 2013) to align the sequences and remove paralogues. Finally, all sequences were compiled together and maximum likelihood estimates of ω (dN∕dS) were calculated using CODEML in PAML 4.9 (Yang, 2007).
To test for selection, we used McDonald-Kreitman (MK) test in DnaSP 6.10.03 to compare species-wide data with outgroup sequences from distantly related Ranidae frogs (Odorrana amamiensis and O. ishikawae, transcriptome data, source: T. Igawa, GenBank accession numbers MH165314 –MH165317). In addition, we tested for sequence-wide neutral (dN = dS), purifying (dN < dS) and positive (dN > dS) selection using codon-based Z tests with 1,000 bootstrap replicates in MEGA7. To infer specific codons as positively selected sites (PSSs) with ω (dN∕dS) > 1, we used omegaMap version 5.0 (Wilson & McVean, 2006) to perform Bayesian inference on independent alignments for each species and gene, following Lau et al. (2016). Neighbour-joining phylogenetic trees from amino acid alignments (p-distance) were constructed independently for TLR2 and TLR4 in MEGA7. Protein domain structures of TLR2 and TLR4 were predicted using SMART (Letunic, Doerks & Bork, 2015).
Expression of TLRs
In order to investigate baseline expression of our candidate TLRs, we extracted expression levels from our transcriptome data set (Lau et al., 2017). RSEM v 1.3.0 (Li & Dewey, 2011) was used to extract trimmed mean log expression ratio, or TMM-normalized values which represent estimated relative RNA production levels (Robinson & Oshlack, 2010), for TLR2 and TLR4 transcripts in each of the 12 samples (Table S1). We compared TMM-normalized values within adults (blood, skin and spleen), using false discovery rate (FDR) cut-off of 0.001, to determine whether expression was ubiquitous. In addition, we checked if expression in tadpoles was different to adults, using stage 24 and stage 29 tadpoles (in R. japonica and R.ornativentris) (Gosner, 1960). In the cases where full-length contigs were not available from low sequence coverage (R. ornativentris TLR2 and TLR4, and R. t. tagoi TLR4), we obtained expression values for each fragmented contig.
Results
We characterized a total of 27 TLR2 and 20 TLR4 alleles from the three study species, all of which were distinctive at the amino-acid level. Rana japonica had seven TLR2 and five TLR4 alleles, R. ornativentris had ten TLR2 and seven TLR4 alleles, and R. tagoi tagoi had ten TLR2 and eight TLR4 alleles (Table 1, Figs. S3 and S4, GenBank accession numbers MG999527–MG999573). All alleles clustered phylogenetically into species-specific clades (Fig. 1) and had similar domain structure to that of other frogs (Fig. S5). Allelic diversity among species was high (Table 1, Table S3), especially TLR2 in R. ornativentris and R. t. tagoi, where all individuals were heterozygous with two unique alleles.
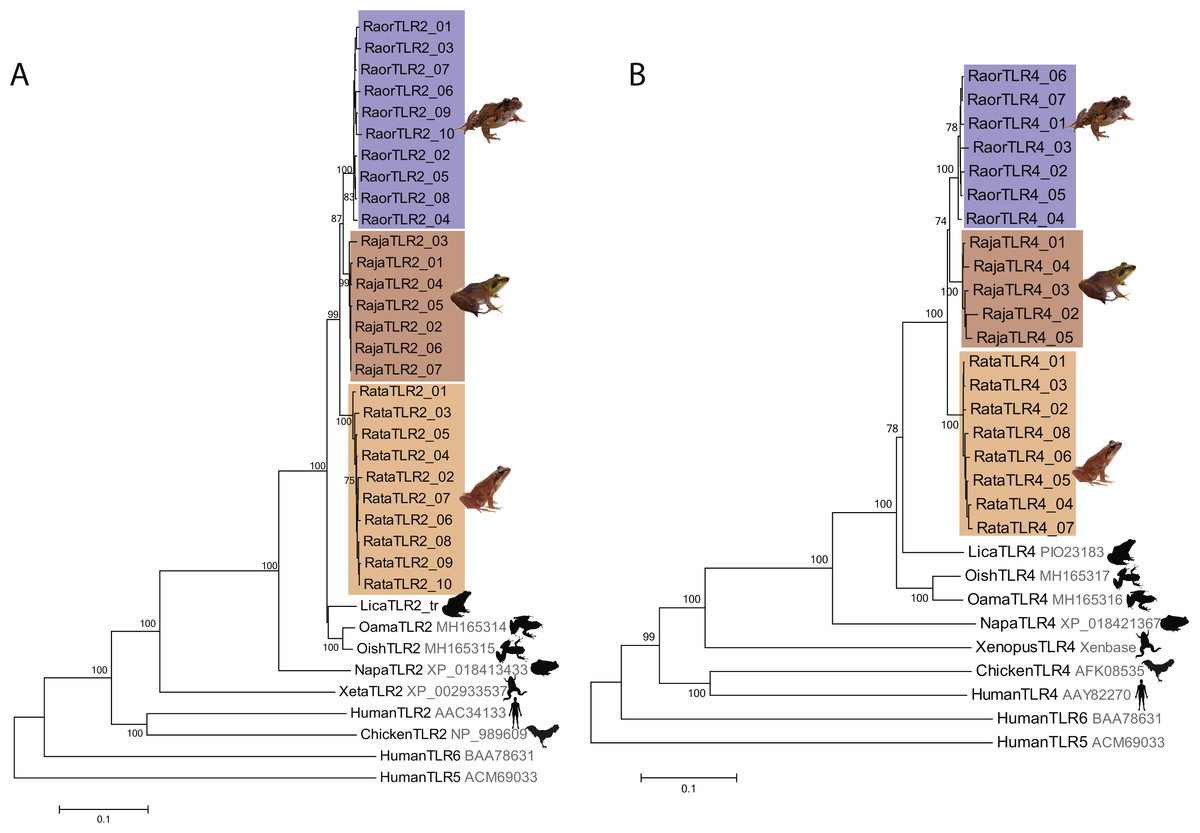
Figure 1: Phylogenetic relationships of TLR2 and TLR4 alleles from three Japanese Rana species.
Phylogenetic relationships of (A) TLR2 and (B) TLR4 alleles identified in R. japonica (red), R. ornativentris (blue) and R. tagoi tagoi (orange) and other species based on amino acid alignments (neighbour-joining method). Human TLR5 and TLR6 were used as outgroup sequences. Accession numbers for sequences are indicated. Sequences obtained from transcriptome data include: Lithobates catesbeianus (DRA accession number SRP051787), and Odorrana amamiensis and O. ishikawae (GenBank accessions MH165314 –MH165317). Image sources: Q. Lau.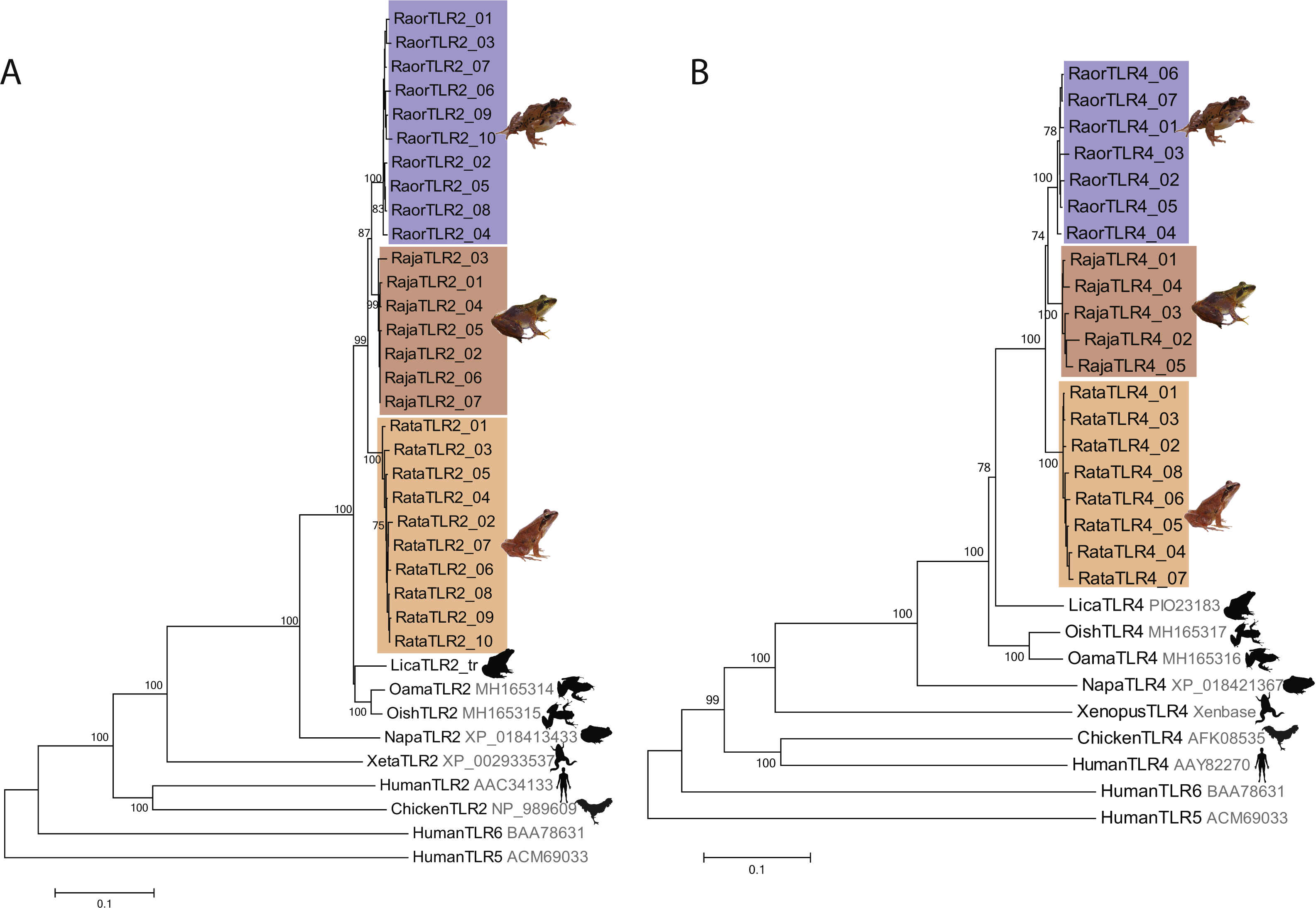{kind=link}
| Gene | 2N | nsites | NA | S | k | π | D | Hn | MK P-value |
|---|---|---|---|---|---|---|---|---|---|
| TLR2 | |||||||||
| R. japonica | 10 | 2,312 | 7 | 28 | 10.48 | 0.0045 | −0.474 | −0.873 | 0.629 |
| R. ornativentris | 10 | 2,312 | 10 | 44 | 13.89 | 0.0060 | −0.521 | −0.242 | 1.000 |
| R. tagoi tagoi | 10 | 2,312 | 10 | 39 | 13.29 | 0.0058 | −0.292 | −1.108 | 0.215 |
| TLR4 | |||||||||
| R. japonica | 10 | 2,072 | 5 | 41 | 19.20 | 0.0093 | −0.183 | −0.144 | 0.279 |
| R. ornativentris | 10 | 2,078 | 7 | 35 | 14.71 | 0.0071 | 0.008 | −0.510 | 0.266 |
| R. tagoi tagoi | 10 | 2,072 | 8 | 25 | 8.57 | 0.0041 | −0.584 | −1.539 | 0.127 |
Notes:
- 2N
-
number of gene copies studied
- nsites
-
nucleotide length of sequence
- NA
-
number of alleles
- S
-
number of segregating sites
- k
-
average number of nucleotide differences
- π
-
nucleotide diversity
- D
-
Tajima’s D value for all sites (no values were significant at p < 0.01)
- Hn
-
Fay and Wu’s normalized H value for all sites (no values significant p < 0.01)
- MK P-value
-
McDonald and Kreitman Fisher’s exact test P-value
Selection tests over the entire alignment indicated that TLR2 and TLR4 in the three focal species are under purifying selection (Z-value = 2.66 − 3.83, p < 0.01, Table 2) with the exception of TLR4 in R. t. tagoi (Z-value = 1.28, p = 0.102). In addition, Tajima’s D, normalized Fay and Wu’s H, and the McDonald-Kreitman test showed no significant support for selection (Table 1). However, omegaMap analyses identified six to nine positively selected sites (PSSs) in either TLR2 or TLR4 of each of the three focal species (Posterior probability >99%). Of these, two (22.2%) to six (75.0%) PSSs were located in predicted leucine rich repeat domains (Table 2, Figs. S3 and S4). There were no PSSs common in all three species studied, but two PSSs of TLR4 (sites D56 and S65) were shared across two species (Table 2, Fig .S4).
| Gene | Neutrality | Purifying | Positive | Positively selected sites (PSS) |
|---|---|---|---|---|
| TLR2 | ||||
| R. japonica | −2.72* | 2.69* | −2.63 n.s. | 12, 23, 95#, 164, 428#, 672 |
| R. ornativentris | −2.78* | 2.66* | −2.77 n.s. | 11, 53#, 75#, 207, 284, 299, 417, 509, 535 |
| R. tagoi tagoi | −3.81** | 3.83** | −3.76 n.s. | 105#, 192, 235, 265, 407, 458#, 485#, 486# |
| TLR4 | ||||
| R. japonica | −3.16* | 3.11* | −3.11 n.s. | 12, 46#, #, 129, 133, 221, 339, 430# |
| R. ornativentris | −3.34* | 3.34** | −3.32 n.s. | 35#, #, 77#, 173, 253#, 378#, 481#, 691 |
| R. tagoi tagoi | −1.28 n.s. | 1.28 n.s. | −1.27 n.s. | 24, 53#, #, #, 127, 128, 373#, 416#, 489# |
Pairwise comparisons of dN∕dS ratios between Rana species were low and ranged from 0.188 to 0.398 (Table 3). These ratios were comparable to that of MHC class 1 α3 and MHC class II β2 domains as well as other transcribed genes within the species (mean dN∕dS of over 3,000 genes = 0.380, Fig. 2). In contrast, these ratios were lower than that of MHC class I α1 and α2 domains and class II β1 domain.
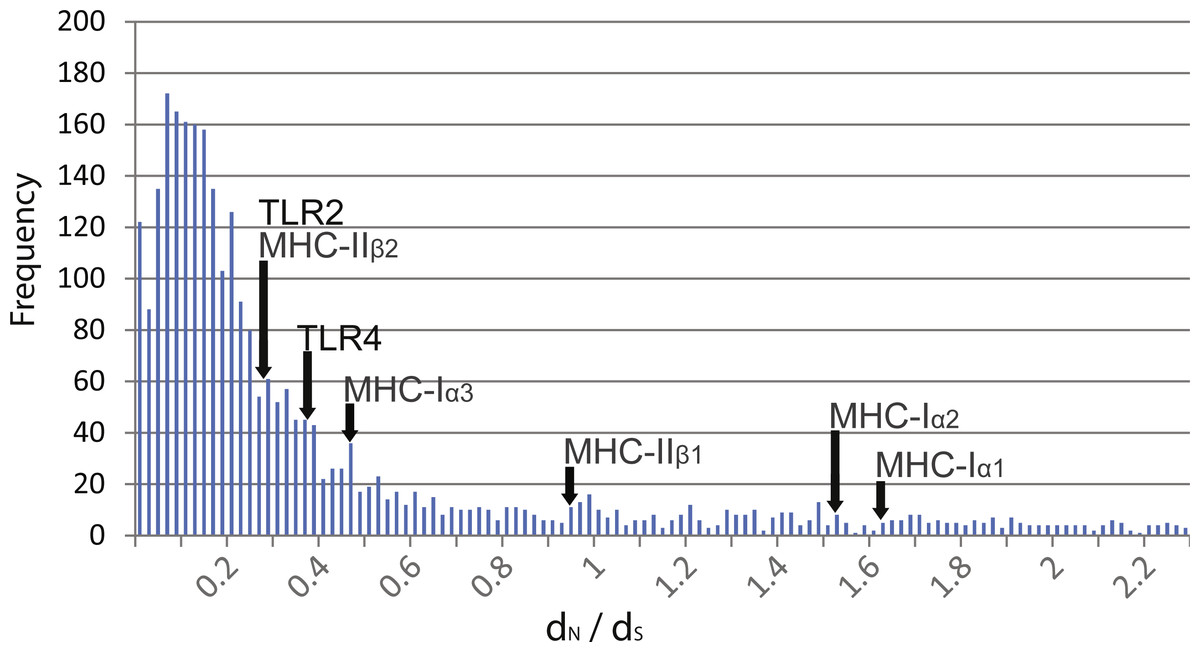
Figure 2: Distribution of dN∕dS ratios from over 3,000 genes isolated from transcriptome data compared to TLR and MHC.
Distribution of dN∕dS ratios from over 3,000 genes isolated from transcriptome data of R. japonica, R. ornativentris and R. tagoi tagoi. dN∕dS location of TLR2 and TLR4, as well as MHC class I and II domains are marked with arrows.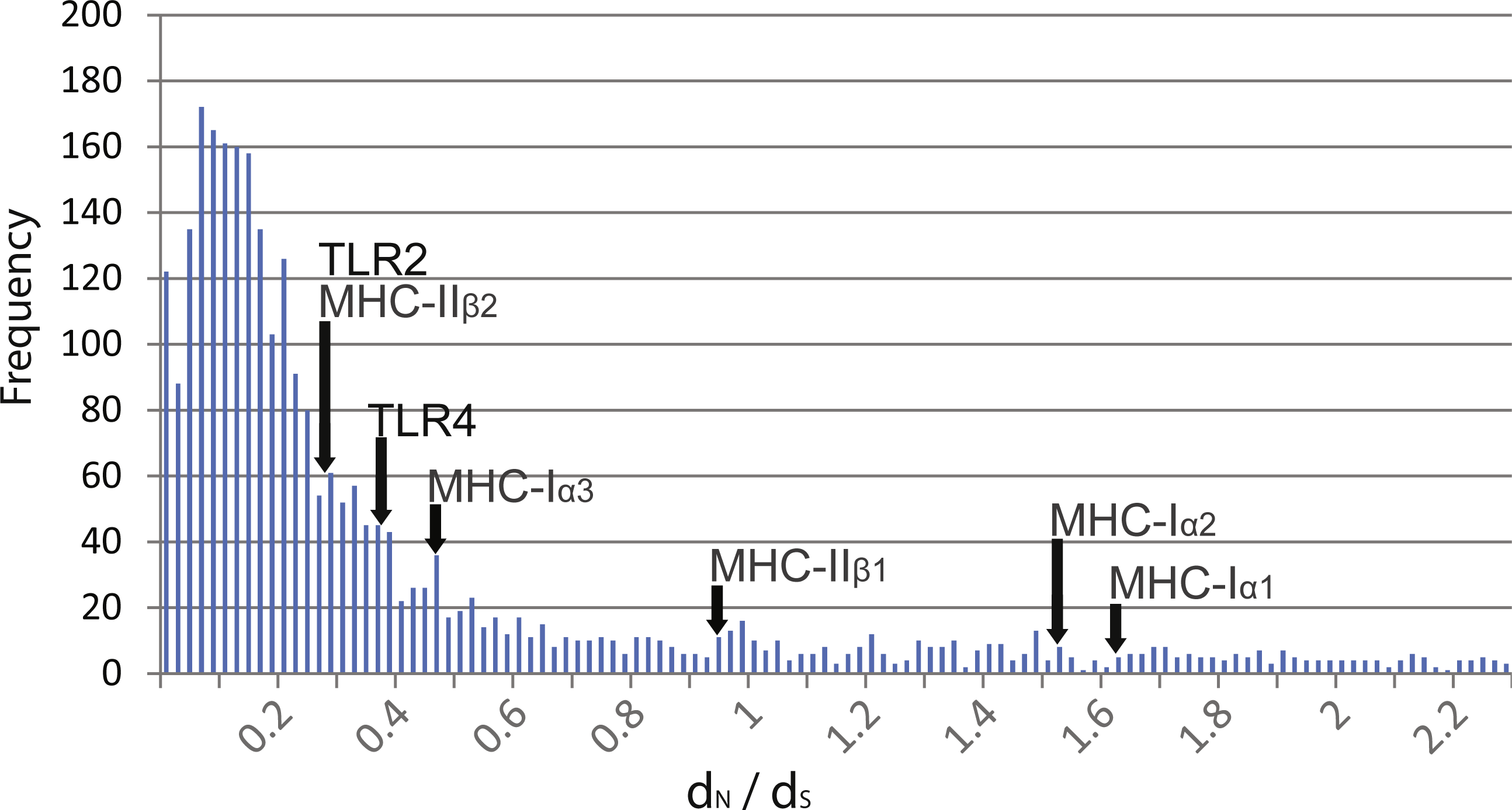{kind=link}
| Gene | dN | dS | dN∕dS |
|---|---|---|---|
| TLR2 | |||
| Rj- Ro | 0.014 | 0.077 | 0.188 |
| Rj-Rt | 0.015 | 0.069 | 0.213 |
| Ro-Rt | 0.020 | 0.065 | 0.316 |
| all three species | 0.291 | ||
| TLR4 | |||
| Rj- Ro | 0.016 | 0.078 | 0.207 |
| Rj-Rt | 0.018 | 0.062 | 0.293 |
| Ro-Rt | 0.020 | 0.051 | 0.398 |
| all three species | 0.379 | ||
| MHC class I α1 | 0.092 | 0.056 | 1.632 |
| MHC class I α2 | 0.082 | 0.054 | 1.525 |
| MHC class I α3 | 0.035 | 0.073 | 0.476 |
| MHC class II β1 | 0.139 | 0.146 | 0.953 |
| MHC class II β2 | 0.041 | 0.151 | 0.269 |
When we checked published transcriptome data for TLR2 and TLR4 expression, preliminary examination of adult tissues indicated no significant differences between tissue types (blood, spleen, skin) at FDR cut-off of 0.0001. Nevertheless, overall expression of TLR2 and TLR4 was low and there were a few consistent trends seen across all three species. This included higher expression of TLR2 in adult skin relative to blood, and lower expression of TLR4 in adult skin relative to either blood or spleen (Table 4). Across life stages, expression of TLR2 and TLR4 was seemingly low in tadpoles relative to adult samples (FDR > 0.0001, n.s., Table 4).
| Gene | Species | Transcript length (bp) | Adult blood | Adult skin | Adult spleen | S24 tadpole | s29 tadpole |
|---|---|---|---|---|---|---|---|
| TLR2 | R.japonica | 3,285 | 0.25 | 1.37 | 2.60 | – | 0.05 |
| R. ornativentris | |||||||
| fragment 1 | 1,417 | 0 | 0.73 | 0.11 | 0.20 | 0 | |
| fragment 2 | 1,324 | 0.16 | 0.88 | 0.22 | 0.25 | 0 | |
| R. t. tagoi | 2,686 | 0.86 | 2.55 | 1.59 | – | – | |
| TLR4 | R.japonica | 2,830 | 0.20 | 0.03 | 1.14 | – | 0.01 |
| R. ornativentris | |||||||
| fragment 1 | 444 | 0.16 | 0 | 0.32 | 0 | 0 | |
| fragment 2 | 548 | 0 | 0 | 0.55 | 0 | 0.52 | |
| fragment 3 | 405 | 0 | 0.28 | 0.58 | 0 | 0 | |
| R. t. tagoi | |||||||
| fragment 1 | 1,454 | 0.11 | 0 | 0.50 | – | – | |
| fragment 2 | 629 | 0.18 | 0 | 1.21 | – | – |
Discussion
The preliminary characterization of TLR2 and TLR4 in the three focal species here provides a platform for future population genetics studies across the species’ distributions, uncovering the full TLR diversity with more targeted PCR and sequencing approaches. Overall allelic diversity of TLR2 and TLR4 appeared to be high, whereby only a few alleles were shared between more than one individual (Table S3). Commonly shared alleles were more apparent in R. japonica, likely due to a population bottleneck history in the source population of Etajima (Lau et al., 2016). The presence of the TLR4 gene in amphibians was previously unclear, described as ‘putative’ in Xenopus frogs (Ishii et al., 2007) and ‘predicted’ from genomic data in Lithobates catesbeianus and Nanorana parkeri (GenBank accessions XP_018421367 and PIO23183; Sun et al., 2015; Hammond et al., 2017), and undetected in newts (Babik et al., 2014). The characterization of TLR4 genes in this study supports the existence of this gene family in anurans, whereby TLR4 alleles of the three Rana species were similar in phylogeny and domain structure to that of other frogs.
From selection tests, we found overall evidence of purifying selection and no support for sequence-wide positive selection. This agrees with data from other vertebrates, including newts, where TLRs are regarded as conserved with their evolution predominated by purifying selection (Roach et al., 2005; Babik et al., 2014). The dN∕dS ratios in TLR2 and TLR4 of the Rana species studied here were remarkably low compared to that of external domains of MHC class I (α1 and α2 domains) and class II (β1 domain), which are considered to be under balancing selection. However, the low dN∕dS of TLR2 and TLR4 was comparable to dN∕dS of MHC class 1 α3 and MHC class II β2 domains which are intracellular or not involved in peptide recognition, as well as those of over 3,000 transcribed genes within the species. These findings further support that TLR2 and TLR4 are under functional constraint.
Although we found that most of the TLR2 and TLR4 sequences of the Japanese Rana frogs were evolutionarily constrained, we identified evidence of adaptive evolution occurring at individual codon sites in our alignment, similar to other vertebrates studied (Wlasiuk & Nachman, 2010; Shang et al., 2018). When comparing with codon sites predicted to be important for binding of non-fungal ligands (Figs. S3 and S4), two PSSs identified in R.ornativentris TLR2 (Q284 and V299) corresponded to sites in human TLR2 predicted to be involved in ligand binding of lipopeptides (N294 and L312, Jin et al., 2007). In addition, one PSS each of R. tagoi tagoi (T128) and R. ornativentris (Q253) corresponded to human TLR4 sites predicted to be involved in secondary (N268) and phosphate (K388) binding, respectively, of bacterial lipopolysaccharides (Park et al., 2009). Positive selection at identical codon sites across different species, as observed in MHC adaptive immune genes (Lau et al., 2016; Lau et al., 2017), could be driven by a single selective force that is pathogen-related. However, in Rana TLRs there were no PSSs shared across all three species studied. As we did not examine the patterns of selection in species which are susceptible to Bd, any link is currently speculative; nevertheless, we cannot rule out the possibility of adaptive evolution, potentially driven by pathogens such as Bd, acting on TLR of the study species in recent evolutionary history.
Preliminary examination of TLR2 and TLR4 expression levels extracted from transcriptome data showed overall low expression. While the expression data is derived from single individuals that were housed in disease-free environments, it appears that skin of healthy frogs that are not immune-challenged express TLR2 more so than TLR4. However, expression of immune-related genes could be modulated following immune or stress challenges, and future studies should monitor immune gene expression following experimental infection with pathogens like Bd. The adult tissue-specific differences in TLR2 and TLR4 expression from R. ornativentris in this study were distinct from B. maxima (Zhao et al., 2014), but sample size should be increased for both species before further inferences can be made. A previous study in Xenopus frogs detected ubiquitous expression of both TLR2 and TLR4 in adults and tadpoles using PCR, but expression levels were not quantified (Ishii et al., 2007). Although we found low TLR expression in tadpoles in this study, further conclusions cannot be made due to limited sampling and overall low TLR expression across the samples. Future quantitative studies can investigate expression level changes of TLRs during development from tadpole to adults, as well as that of other innate and adaptive immune genes extracted from the transcriptome data set (Lau et al., 2017).
Conclusion
In this study, we characterized TLR2 and TLR4 genes from three Japanese Rana species. We provide strong evidence of purifying selection acting across TLR2 and TLR4, and evidence of a few specific codon sites under positive selection. Further research is necessary to determine if the positive selection we detected is due to pathogen-driven selection. Since immunity to infectious diseases is usually polygenetic, our study adds to the growing body of literature related to genes that potentially impact resistance to Bd and other pathogens in amphibians.